
Nicht-dystrophe Myotonie und periodische Paralyse im Kindesalter: behandelbare muskuläre Kanalopathien
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Nicht-dystrophe Myotonien (NDM) stellen die größte Untergruppe der muskulären Kanalopathien dar. Die Behandlung dieser Erkrankungsgruppe gelingt im Kindesalter meist mit zufriedenstellender Symptomkontrolle. Die sehr seltene periodische Paralyse (PP) als weitere Untergruppe der muskulären Kanalopathien stellt klinisch oft eine Mischform zwischen Myotonie und Paralyse dar. Umgekehrt gehen einige Unterformen der NDM mit PP einher.
Keypoints
-
NDM sind seltene, gut behandelbare genetische Erkrankungen. Die Lebenserwartung ist in aller Regel nicht vermindert.
-
Trigger wie Kälte, Schwangerschaft, Narkosen sowie Übergänge zwischen Ruhe und Bewegung (und umgekehrt), teilweise auch Hypo- bzw. Hyperkaliämie sollten berücksichtigt werden.
-
Medikamentös lässt sich eine meist befriedigende Symptomkontrolle erzielen, wobei bei unbefriedigendem Ansprechen mehrere Substanzen versucht werden sollten.
-
Empfohlen werden regelmäßige EKG-Kontrollen, da eine erhöhte Neigung zu Herzrhythmusstörungen besteht.
Myotonie beschreibt eine verzögerte Entspannung der Skelettmuskulatur nach einer willkürlichen Kontraktion und wird von Patient:innen als Gefühl der Muskelsteifheit wahrgenommen. Gelegentlich geht dieses Gefühl einher mit Schmerzen, Müdigkeit und Schwäche. Am charakteristischsten ist die Handmyotonie, das verzögerte Öffnen der Hand nach kräftigem Faustschluss. Daneben sind myotone Reaktionen an praktisch allen Skelettmuskeln möglich, sie betreffen je nach Krankheitsentität vorrangig die Oberschenkel- oder die Gesichtsmuskeln.
Der Pathomechanismus der myotonen Reaktion ist ein gestörtes Gleichgewicht von Depolarisation und Repolarisation der Muskelzellen, dessen Resultat eine verlängerte Depolarisation der Zelle ist (Abb. 1). Im Falle der CLCN1-Mutation („loss of function“ [LOF]) liegt eine fehlende Repolarisation der Chloridkanäle an der Muskelzelle zugrunde, bei SCN4A-Mutationen („gain of function“ [GOF]) hingegen eine exzessive Depolarisation der Kaliumkanäle. Die periodische Paralyse (PP) führt zu episodischer schlaffer Lähmung eines Großteils der Skelettmuskulatur, wobei zwischen den Anfällen meist normale Muskelkraft besteht. Bekannte Ursachen sind Mutationen im SCN4A-, CACNA1S- oder KCNJ2-Gen. Mechanistisch kommt es durch verlängerte Inaktivierung zu einer Nicht-Erregbarkeit der Muskelzelle. 1

Abb. 1: Überblick über die Erregungs-Kontraktions-Kopplung im normalen Skelettmuskel. Nach der Auslösung des Aktionspotenzials durch ein Motoneuron breitet sich das Aktionspotenzial (das von nahe gelegenen spannungsgesteuerten Natriumkanälen des Skelettmuskels, Nav1.4, erzeugt wird) entlang des Sarkolemms in die T-Tubuli aus. Hier öffnen spezielle spannungsgesteuerte Kalziumkanäle (Dihydropyridinrezeptoren, DHPR) die Ryanodinrezeptoren (RyR) und aktivieren diese, die ihrerseits Kalzium aus dem sarkoplasmatischen Retikulum (SER) ins Zytoplasma freisetzen und so die Muskelkontraktion auslösen. Chloridkanäle (ClC-1) üben durch den Einstrom von Chloridanionen eine stabilisierende Wirkung auf die Membran der ruhenden Skelettmuskelzelle aus; während der Depolarisation werden ClC-1 noch stärker aktiviert und tragen dazu bei, die Zellmembran nach einem Aktionspotenzial zu repolarisieren (nach Bandschapp O et al. 2013)1
Die muskulären Kanalopathien werden im Gegensatz zur myotonen Dystrophie Typ 1 und Typ 2 (DM1 und DM2) als nichtdystrophe Myotonien (NDM) bezeichnet. Sie umfassen eine Gruppe seltener, gutartiger, genetisch bedingter Muskelkanalopathien, deren Hauptsymptom die Skelettmuskel-Myotonie darstellt. In Abgrenzung hierzu stellen DM1 und DM2 systemische neuromuskuläre Erkrankungen dar, bei denen Myotonie mit einer progredienten Myopathie und multisystemischen Manifestationen vergesellschaftet ist.
NDM sind selten, je nach Studie liegt ihre Prävalenz bei ca. 2–2,4 pro 100000 Personen. Gelegentlich führen Founder-Mutationen zum Gendrift, verbunden mit einer deutlich höheren Prävalenz in bestimmten Populationen.2,3
Diagnostik
Im Kindesalter stehen klinische Untersuchung und gründliche Anamnese an erster Stelle. Nicht selten erfährt man hier bereits von einer langjährig vorbestehenden Handmyotonie, welche nicht zum Arztkontakt führte und in der Familie als „normal“ eingeordnet wird (eigene Beobachtung). Bei deutlich nachweisbarer Myotonie empfiehlt sich im Kindesalter im nächsten Schritt eine molekulargenetische Untersuchung, die die Repeat-Expansion-Erkrankungen DM1 sowie selten DM2 miterfassen sollte. Laborchemisch sollte eine Hypo- bzw. Hyperkaliämie ausgeschlossen und eine gelegentliche CK-Erhöhung erfasst werden. Ebenfalls auszuschließen ist eine Hyperthyreose. Aufgrund des seltenen Vorkommens von Arrhythmien bei NDM sollte, insbesondere im Kontext einer Medikation mit Antiarrhythmika, als Ausgangsbefund ein EKG erhoben werden; im Weiteren sind ebenfalls regelmäßige EKG-Kontrollen angezeigt.4
Klassifikation der muskulären Kanalopathien mit wichtigen Merkmalen
Der Krankheitsname orientiert sich sowohl an der klinischen Symptomatik als auch an der zugrunde liegenden Ionenkanalmutation, wie Abbildung 2 verdeutlicht.5
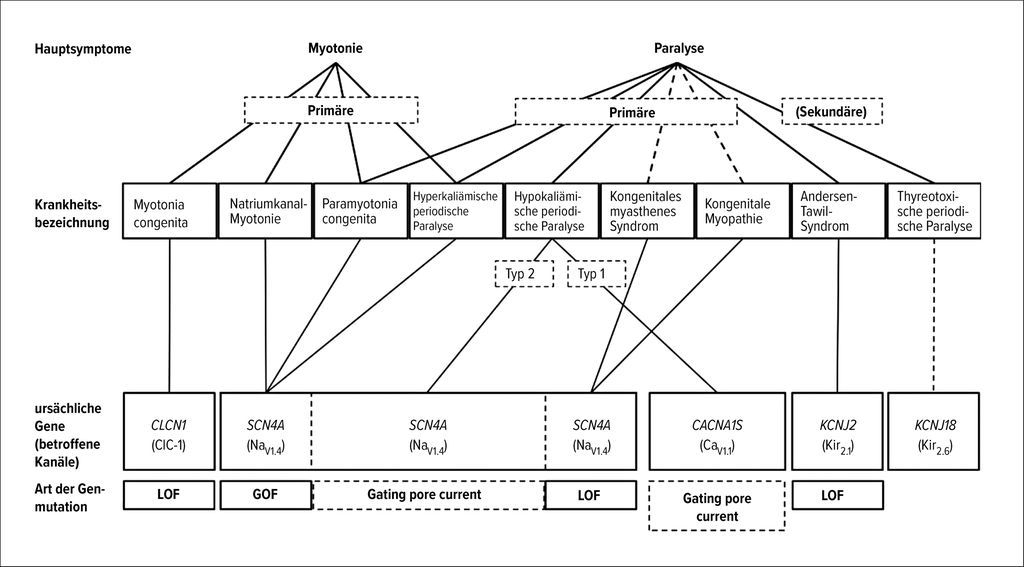
Abb. 2: Überblick über verschiedene nicht-dystrophe Myotonien im Kindesalter und deren genetische Grundlage (nach Kubota, T et al. 2025)5
Myotonien
Hauptmerkmal der Myotonia congenita (MC) und Natriumkanal-Myotonie („sodium channel myotonia“, SCM) ist das Warm-up-Phänomen, bei dem durch wiederholte Anspannung einer Muskelgruppe eine Verbesserung der Myotonie eintritt. Der Erkrankungsbeginn liegt in der 1.–2. Lebensdekade.6 Bei der SCM ist eine faziale Beteiligung mit Myotonie der Augenlider und bulbären Muskeln charakteristisch.4 Bei der seltenen Paramyotonia congenita (PC) verschlechtert sich die Myotonie durch körperliche Betätigung. Die meisten SCN4A-assoziierten Muskelkanalopathien gehen sowohl mit Myotonie als auch Schwäche (bis hin zu PP) einher.
In der Neonatalperiode findet sich als sehr seltenes Krankheitsbild der „severe neonatal episodic laryngospasm“ (SNEL), bei dem es infolge einer Mutation im SCN4A-Gen zu episodischem Laryngospasmus oder Apnoen in den ersten Lebenstagen kommt. Die Ausprägung der Erkrankung ist sehr variabel und geht mit der Zeit oft in eine Myotonia permanens über.8
Periodische Paralyse (PP)
Die PP wird unterteilt in hyperkaliämische PP(Hyper-PP)- und hypokaliämische PP(Hypo-PP)-Formen sowie in das Andersen-Tawil-Syndrom (ATS). Auch bei PP sind SCN4A-Mutationen zu einem Großteil verursachend (bei Hyper- als auch Hypo-PP). Mutationen im Gen des Kalziumkanals (CACNA1S) sind gleichfalls verursachend für Hypo-PP und gehen mit einer signifikanten Hypokaliämie während einer Episode der PP einher. Bei der Hyper-PP ist Kalium hingegen erhöht, diese Erkrankung zeigt interiktal oft auch myotone Entladungen. Das ATS entsteht durch Mutationen im Kaliumkanal-Gen (KCNJ2). Die Tatsache, dass Kaliumkanäle neben Skelettmuskeln auch in anderen Geweben exprimiert sind, erklärt das systemische Krankheitsbild, bei dem auch kardiale und zentrale Symptome vorkommen.
Trigger
-
Kälte aggraviert die Symptome bei allen NDM.7
-
Für alle Muskelkanalkrankheiten ist bei Anästhesien Vorsicht geboten. Succinylcholine und depolarisierende Muskelrelaxanzien können lebensbedrohliche myotone Reaktionen auslösen, der Einsatz nichtdepolarisierender Muskelrelaxanzien sollte vorsichtig titriert werden. Zu achten ist ferner auf Normothermie, zudem bei der CACNA1S-assoziierten Hypo-PP auf die Gefahr einer malignen Hyperthermie.1
-
Auslöser für einen Anfall bei allen Formen der PP sind übermäßige oder ungewohnte körperliche Betätigung, gefolgt von völliger Ruhe. Daneben sind Stress, Schlafmangel, Kälte und Schwangerschaft bekannte Trigger. Fasten und die Aufnahme kaliumreicher Lebensmittel gelten als Auslöser für Hyper-PP, kohlenhydratreiche Lebensmittel für Hypo-PP.9
Therapieoptionen
Bei Myotonien
Therapeutisch haben sich bei Kindern Natriumkanalblocker in der Behandlung der MC bewährt. Mexiletin, ein Antiarrhythmikum der Klasse IB, führt zur schnellen Inaktivierung der Natriumkanäle. Für Erwachsene hat das Präparat Namuscla 2018 als Orphan-Drug die europäische Marktzulassung erhalten. In zwei unabhängigen randomisierten placebokontrollierten Studien zeigte sich eine gute Wirksamkeit zur Symptomkontrolle von Myotonie, Schwäche, Müdigkeit und Schmerzen (Namuscla 167mg, 1–3 Tabletten/Tag). Regelmäßige EKG-Kontrollen vor und nach jeder Dosissteigerung sind beim Off-Label-Gebrauch im Kindesalter zu empfehlen. Mehr Expertise besteht in der Pädiatrie mit dem aus der Epileptologie bekannten Lamotrigin, einem weiteren Natriumkanalblocker. Dieses zeigte in einer randomisierten, placebokontrollierten Studie bei Erwachsenen eine Verringerung der Myotonie (Startdosis 25mg/d mit langsamer Steigerung bis 3x 100mg/d).
Weitere interessante Therapieansätze beinhalten eine Blockade der langsam inaktivierenden SCN4A-Kanäle, entsprechende Substanzen sind Lacosamid, Rufinamid oder Ranolazin.9,10
In einem Case Report zeigte Flecainid bei einem Neugeborenen mit SNEL und heterozygoter Mutation p.G1306E bei einer Dosis von 100mg/d einen zufriedenstellenden Therapieerfolg. Alternativ findet sich in Einzelberichten gutes Ansprechen auf Carbamazepin, Mexiletin oder Acetazolamid bei SNEL.8
Bei Myotonien und PP
Medikamente zur Erhöhung der Chloridleitfähigkeit zur Behandlung der MC stehen nicht zur Verfügung, jedoch fördert Acetazolamid die Öffnung von Chloridkanälen durch Hemmung der Carboanhydrase und intrazellulärer Ansäuerung.11 Es zeigt ein besonders gutes Ansprechen bei SCM, PMC und allen Unterformen der PP. Aufgrund der Gefahr einer durch Acetazolamid verursachten Urolithiasis werden regelmäßige Ultraschalluntersuchungen empfohlen.12 Dichlorphenamid ist ein 30-fach stärkerer Carboanhydrase-Inhibitor als Acetazolamid. Er wird oft als Mittel erster Wahl zur Reduktion paralytischer Attacken bei Hypo-PP (50–0–50mg/d) eingesetzt.13
Literatur:
1 Bandschapp O, Iaizzo PA: Pathophysiologic and anesthetic considerations for patients with myotonia congenita or periodic paralyses. Paediatr Anaesth 2013; 23(9): 824-33 2 Vivekanandam V et al.: Prevalence of genetically confirmed skeletal muscle channelopathies in the era of next generation sequencing. Neuromuscul Disord 2023; 33(3): 270-27 3 Stunnenberg BC et al.: Prevalence and mutation spectrum of skeletal muscle channelopathies in the Netherlands. Neuromuscul Disord 2018; 28(5): 402-7 4 Vereb N et al.: Non-dystrophic myotonias: clinical and mutation spectrum of 70 German patients. J Neurol 2021; 268(5): 1708-20 5 Kubota T, Takahashi MP: Molecular genetics of skeletal muscle channelopathies. J Hum Genet 2025; doi: 10.1038/s10038-025-01370-w 6 Lyons MJ et al.: Novel CLCN1 mutation in carbamazepine-responsive myotonia congenita. Pediatr Neurol 2010; 42(5): 365-8 7 Fournier E et al.: Cold extends electromyography distinction between ion channel mutations causing myotonia. Ann Neurol 2006; 60(3): 356-65 8 Portaro S et al.: Flecainide-responsive Myotonia permanens with SNEL onset: A new case and literature review. Pediatrics 2016; 137(4): e20153289 9 Jitpimolmard N et al.: Treatment updates for neuromuscular channelopathies. Curr Treat Options Neurol 2020; 22(10): 34 10 Novak KR et al.: Sodium channel slow inactivation as a therapeutic target for Myotonia congenita. Ann Neurol 2015; 77(2): 320-32 11 Ruff, RL: Sour on the inside, calm on the outside: how acetazolamide may stabilize membrane excitability. Muscle Nerve 2006; 34(3): 263-4 12 Suetterlin KJ et al.: Annual renal ultrasound may prevent acute presentation with acetazolamide-associated urolithiasis. Neurol Clin Pract 2021: 11(1): e40-e42 13 Sansone VA et al.: Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology 2016; 86(15): 1408-16
Das könnte Sie auch interessieren:

Schlafapnoe bei Frauen
Die obstruktive Schlafapnoe (OSA) wurde über Jahrzehnte als Krankheit von „übergewichtigen, schnarchenden Männern“ simplifiziert. Neue wissenschaftliche Untersuchungen zeigen zunehmend ...

Der Stellenwert von Biomarkern im Zeitalter zielgerichteter Migränetherapien
Die Migränetherapie hat sich in den vergangenen Jahren in Richtung einer spezifischen und pathophysiologisch informierten Behandlung entwickelt. Viele klinische Entscheidungen erfolgen ...

Migräne im Kindes- und Jugendalter: erkennen, verstehen, behandeln
Migräne zählt zu den häufigsten neurologischen Erkrankungen im Kindes- und Jugendalter, wird jedoch im klinischen Alltag häufig übersehen oder fehldiagnostiziert. Die klinische ...