Stellenwert kutaner Leitsymptome im Zeitalter der Molekulargenetik
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Eine schnelle Diagnosestellung bei Kindern mit Genodermatosen ist wesentlich. Hierfür sind molekulargenetische Analysen sehr hilfreich. Um deren Potenzial auszuschöpfen, bedarf es einer genauen Phänotypisierung der Patienten. Der nachfolgende Artikel gibt einen Überblick über kutane Leitsymptome, die auch im Zeitalter der Molekulargenetik einen hohen Stellenwert in der Diagnostik seltener Hautkrankheiten einnehmen.
Keypoints
-
Eine korrekte und schnelle Diagnose ist bei Kindern mit seltenen Hautkrankheiten wichtig.
-
Molekulargenetische Analysen sind bei der Diagnose von Genodermatosen State of the Art.
-
Eine Phänotypisierung ist essenziell, um das Potenzial der Exomsequenzierung und Panelanalyse bestmöglich ausschöpfen zu können.
-
Kutane Leitsymptome haben einen hohen Stellenwert in der Diagnostik seltener Hautkrankheiten.
Bei Kindern mit seltenen Hautkrankheiten (Genodermatosen) muss eine korrekte und schnelle Diagnose angestrebt werden, um eine Verunsicherung der Eltern und gut gemeinte falsche Ratschläge zu vermeiden. Die Diagnose bildet die Grundlage für Informationen über Prognose und Verlauf der Erkrankung, für weiterführende Untersuchungen, die gezielte humangenetische Beratung sowie Kostenersatz für Medikamente und Bewilligung von Pflegegeld. Auch für zielgerichtete Therapien, frühe Förderung bei Entwicklungsverzögerung sowie die Teilnahme an krankheitsspezifischen Studien und Selbsthilfegruppen ist die Diagnosestellung wesentlich.
Phänotypisierung und molekulargenetische Analyse
Genodermatosen werden heutzutage überwiegend nach genauer klinischer Untersuchung (Phänotypisierung) mithilfe molekulargenetischer Analysetechniken diagnostiziert. Daneben spielen je nach Hautkrankheit auch (Familien-)Anamnese, Labordiagnostik, Histologie, Immunfluoreszenz, Elektronenmikroskopie und bildgebende Untersuchungen eine ergänzende Rolle.
Die derzeit überwiegend in der Diagnostik verwendeten molekulargenetischen Analysetechniken umfassen die Panel-Diagnostik, bei der Exons ausgewählter Gene ausgewertet werden, und die Exomsequenzierung („whole exome sequencing“, WES), bei der alle bekannten Exons und einzelne regulatorische Squenzen (ca. 1% des Genoms) untersucht werden. Die Etablierung dieser Sequenziertechniken hat den Vorteil, dass dadurch andere invasive Diagnosemethoden ersetzt werden können. Bei einem Kollodiumbaby z.B. ist meist keine Hautbiopsie mehr erforderlich. Auch für die Genotyp-Phänotyp-Korrelierung ist eine molekulargenetische Untersuchung Voraussetzung.
Nachteile stellen die Auswertung großer Sequenz-Datenmengen und die nicht immer einfache Klassifizierung der detektierten Varianten dar. Beispielsweise muss mithilfe spezieller Computerprogramme und (hauseigener) Datenbanken zwischen krankheitsursächlicher Mutation, „single nucleotid polymorphism“ (SNP) und „Variante von unbekannter Signifikanz“ (VUS) differenziert werden.
Eine Genpanel-Analyse (ca. 20 Gene) kostet ca. 2100 Euro und benötigt in etwa 6 Wochen. Wichtig ist, dass WES und Panel-Diagnostik keinesfalls eine dezidierte klinische Untersuchung des betroffenen Kindes ersetzen. Um das Potenzial der WES auszuschöpfen, bedarf es einer umfangreichen Phänotypisierung. Kutane Leitsymptome haben daher nach wie vor einen hohen Stellenwert in der Diagnostik seltener Hautkrankheiten. Sie müssen jedoch erkannt werden! Tabelle 1 und 2 fassen einige hilfreiche Leitsymptome der Haut und anderer ektodermaler Strukturen zur Diagnose von Genodermatosen zusammen.
Im Folgenden möchte ich etwas näher auf das Leitsymptom Flecken eingehen, weil in der Praxis relativ häufig Kinder mit hyper- oder hypopigmentierten Maculae an unsere Genodermatosen-Sprechstunde überwiesen werden.
Leitsymptom Café-au-lait-Flecken vs. Eschenlaubflecken
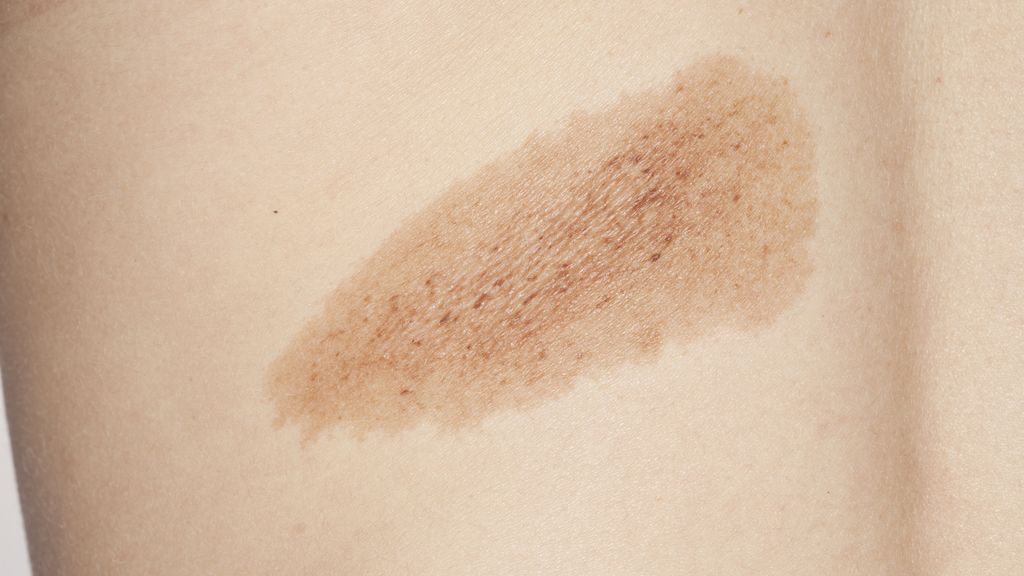
Die häufig schon bei der Geburt vorhandenen Café-au-lait-Flecken zeichnen sich durch eine braune homogene Pigmentierung und rundliche bis ovale Grundform mit scharfer Begrenzung aus (Abb. 1A). Sie sind nicht erhaben und regellos über den Körper verteilt, kommen jedoch palmoplantar nicht vor. Anzahl und Größe der Läsionen sind variabel, Letztere beträgt wenige Millimeter bis über 20 Zentimeter. Histologisch findet sich in den Hyperpigmentierungen eine Vermehrung von Melanozyten.
Pigmentflecken haben in geringer Anzahl keinen Krankheitswert, bis zu 20% der Normalbevölkerung weisen einen bis wenige Flecken auf. Ein gehäuftes Auftreten findet sich bei hereditären neurokutanen Erkrankungen wie beispielsweise Neurofibromatose Typ 1 (Gen: NF1), McCune-Albright-Syndrom („guanine nucleotide binding protein [G protein], alpha stimulating activity polypeptide 1“; GNAS1) oder Noonan-Syndrom (50% der Patienten zeigen Mutationen in PTPN11 [„protein tyrosine phosphatase non-receptor type 11“]) (Tab. 1).
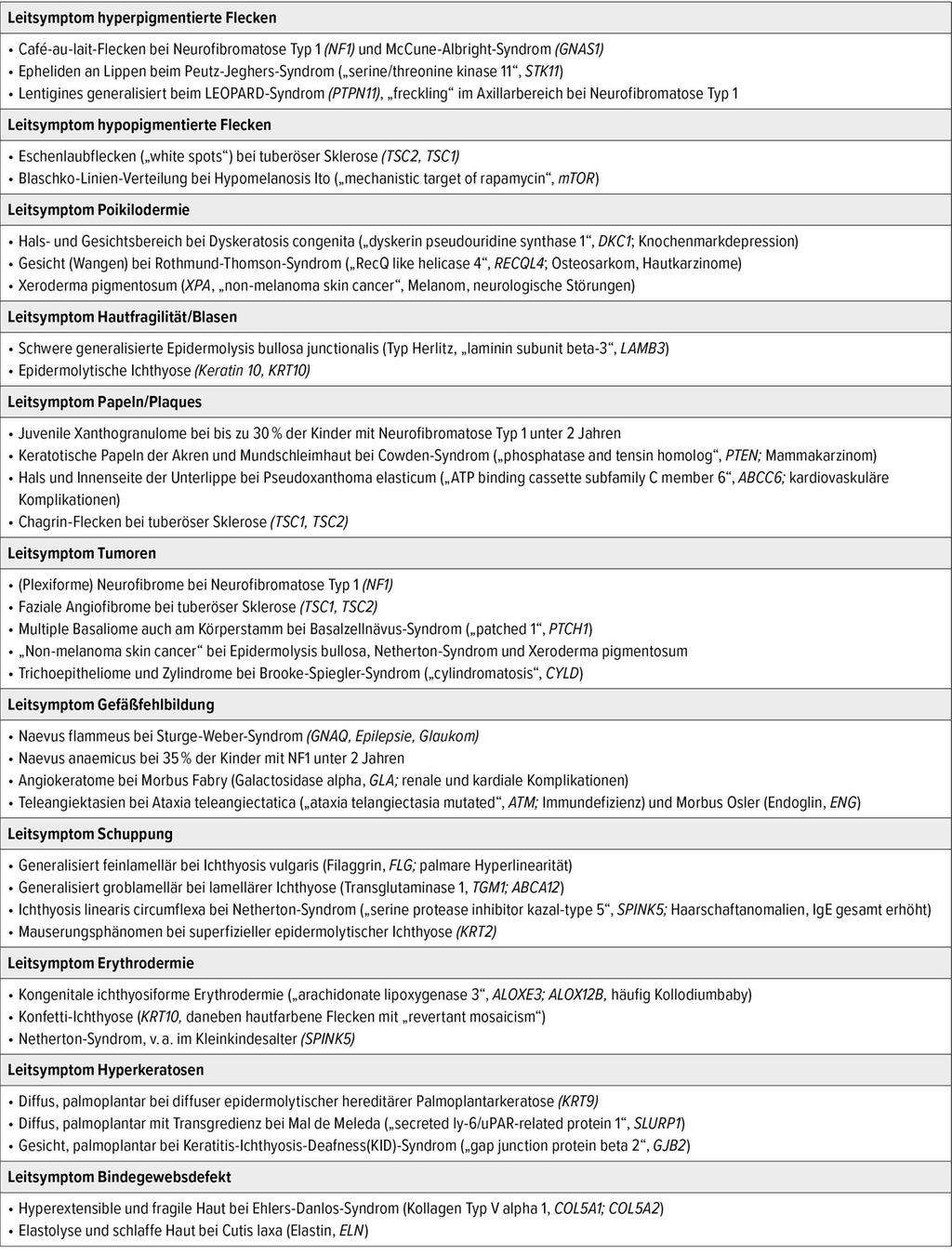
Tab. 1: Leitsymptome an der Haut für die Diagnose von Genodermatosen (Auswahl)
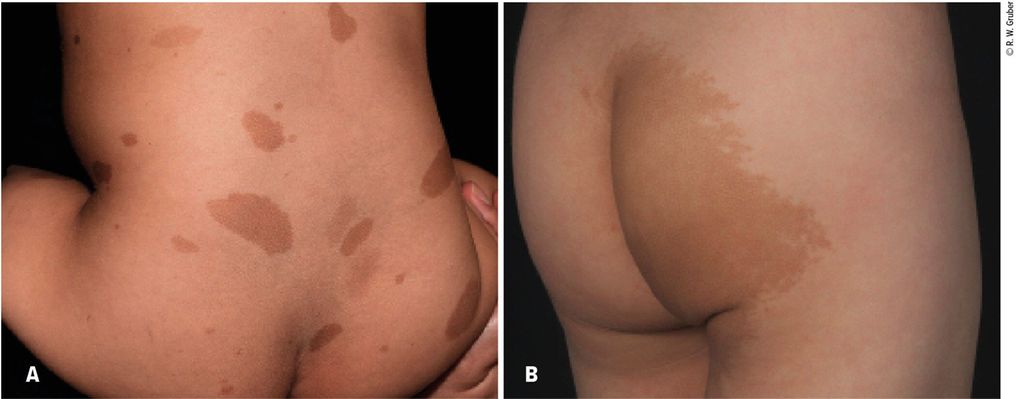
Abb. 1: Morphologisch unterschiedliche Café-au-lait-Flecken bei je einem Kind mit Neurofibromatose Typ 1 (A) und McCune-Albright-Syndrom (B)
Sechs oder mehr Café-au-lait-Flecken von über 5mm Durchmesser im Kindesalter bzw. über 15mm Durchmesser im Erwachsenenalter gelten als Diagnosekriterium der Neurofibromatose Typ 1. Mehr als 95% der Patienten mit molekulargenetisch gesicherter Neurofibromatose Typ1 weisen diese charakteristischen Hyperpigmentierungen an der Haut auf. Typischerweise zeigen Café-au-lait-Flecken bei der Neurofibromatose eine runde Begrenzung, was mit der geradlinigen Küste Kaliforniens („coast of California“) verglichen wird, und sind wenige Zentimeter groß (Abb. 1A). Im Vergleich dazu sind Café-au-lait-Flecken beim McCune-Albright-Syndrom meist unilateral lokalisiert, mehrere Zentimeter groß und weisen eine irreguläre Begrenzung mit gezacktem Rand auf, was auf einer Landkarte an die zerklüftete und inselreiche Küste von Maine („coast of Maine“) erinnert (Abb. 1B).
Bis zu 80% der Kinder mit Neurofibromatose Typ 1 zeigen überwiegend axillär und inguinal sommersprossenartige Hyperpigmentierungen, was als „freckling“ bezeichnet wird und ein weiteres Diagnosekriterium darstellt. Andere hilfreiche Leitsymptome für die Diagnose Neurofibromatose Typ 1 bei Kleinkindern inkludieren juvenile Xanthogranulome, die bei bis zu 30% der Patienten unter zwei Jahren gefunden werden, sowie den Naevus anaemicus, der bei 35% der betroffenen unter Zweijährigen vorkommt (Tab. 1).
Bei den Eschenlaubflecken („white spots“) handelt es sich um kongenitale hypopigmentierte Flecken von meist länglicher, blattförmiger Grundform mit scharfer Begrenzung (Abb. 2). Sie sind bis zu einige Zentimeter groß und regellos über den Körper verteilt. Histologisch finden sich in den hypomelanotischen Flecken zwar Melanozyten, diese produzieren aber weniger Melanosomen. Einzelne „white spots“ (Naevus depigmentosus) kommen bei bis zu 4% gesunder Neugeborener vor, das Vorliegen von mindestens drei Eschenlaubflecken ist jedoch ein Hauptkriterium für die Diagnose einer tuberösen Sklerose (TSC1, TSC2)(Tab. 1).
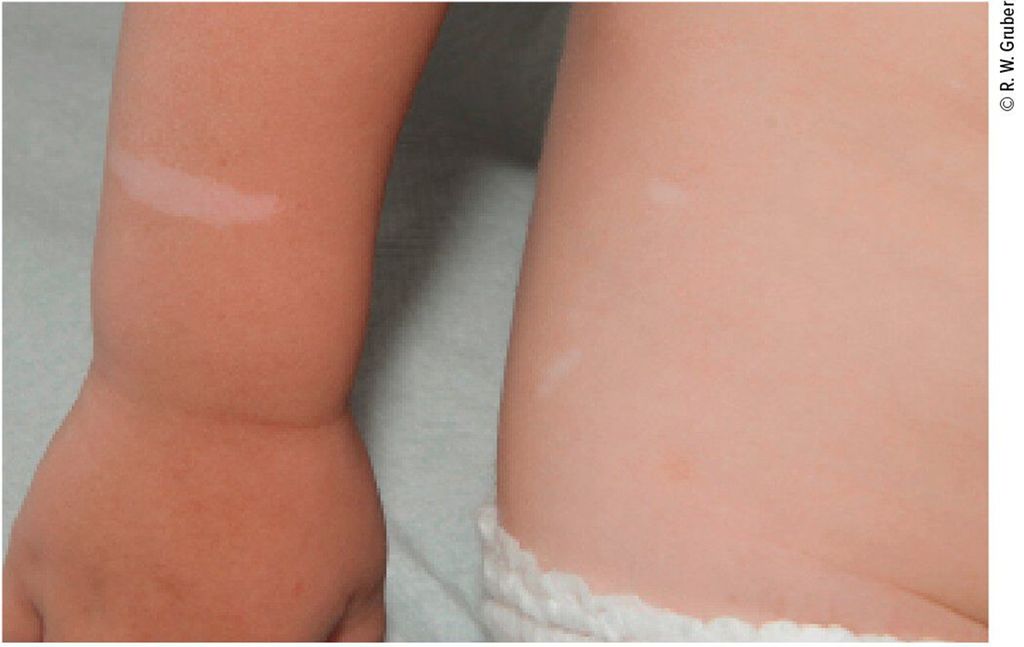
Abb. 2: Typische Eschenlaubflecken bei einem Kind mit tuberöser Sklerose
Ein Teil der Kinder mit tuberöser Sklerose präsentiert auch für die Erkrankung charakteristische kleine, wenige Millimeter große, hypopigmentierte Konfetti-Flecken, die vor allem an den Extremitäten vorkommen. Die hypopigmentierten Flecken werden üblicherweise mit bloßem Auge erkannt, bei Kindern mit hellem Hauttyp können diese jedoch leicht übersehen werden. Bei der klinischen Untersuchung ist daher die Verwendung einer Wood-Lampe hilfreich. Bei fehlender Verfügbarkeit einer Wood-Lampe kann alternativ auch das blaue Licht eines Smartphones, das durch ein homogenblaues Bild am Screen erzeugt wird, für die Visualisierung der Hypopigmentierungen verwendet werden.
Die Diagnose nicht unbedingt vereinfachend ist die Tatsache, dass bis zu einem Drittel der Kinder mit tuberöser Sklerose neben den typischen Eschenlaubflecken auch einige wenige Café-au-lait-Flecken aufweist. Allgemein sollte bei Kindern, die multiple hyperpigmentierte oder hypopigmentierte Maculae zeigen, unbedingt eine Genodermatose ausgeschlossen werden.
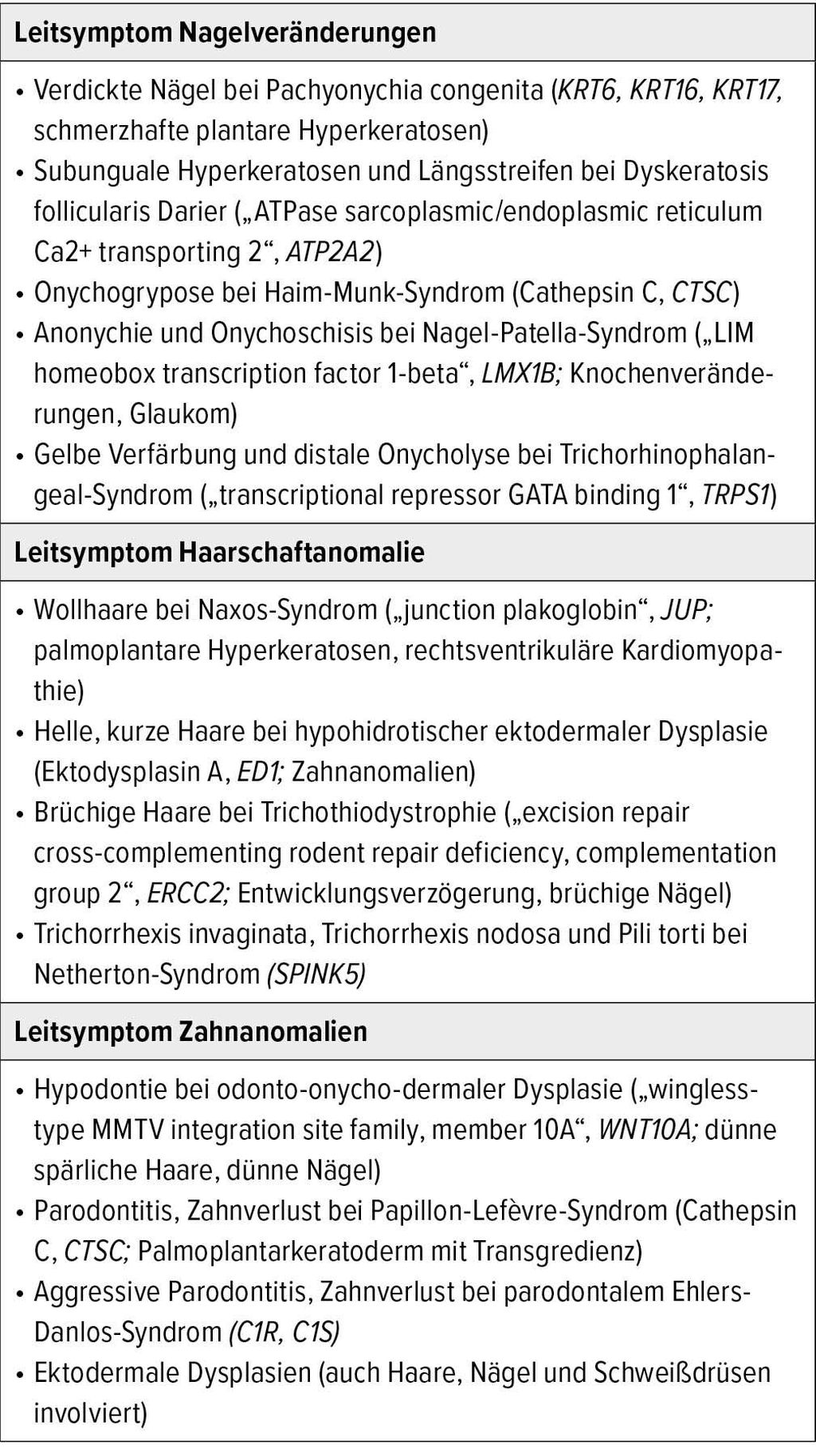
Tab. 2: Leitsymptome anderer ektodermaler Strukturen für die Diagnose von Genodermatosen (Auswahl)
Zusammenfassend kann man festhalten, dass meist mithilfe molekulargenetischer Analysen, für welche eine genaue Phänotypisierung der Patienten eine wesentliche Voraussetzung darstellt, eine schnelle Diagnose bei Kindern mit seltenen Hautkrankheiten möglich ist. Kutane Leitsymptome haben dabei einen hohen Stellenwert.
Literatur:
beim Verfasser
Das könnte Sie auch interessieren:

Tägliche Tablette gegen Psoriasis
Die US-Arzneimittelbehörde FDA hat mit Icotrokinra ein orales Medikament gegen Schuppenflechte zugelassen, welches die Rezeptoren für Interleukin-23 (IL-23) hemmt. Eine EU-Zulassung ...

Aminosäuren – Booster für die Wundheilung?
Für den Wundheilungsprozess ist je nach Heilungsprozess die richtige Kombination aus Kohlenhydraten, Fetten und Proteinen sowie aus Mineralien, Spurenelementen und Vitaminen essenziell. ...

Ein haariger Fall mit irreversiblen Folgen
Bestimmte Formen von Alopezie scheinen in jüngster Zeit explosionsartig zuzunehmen, wobei die genauen Ursachen bislang noch nicht vollständig geklärt sind. Handelt es sich dabei um eine ...