
Les limites de l’échelle ALSFRS-R en tant qu’indicateur de l’évolution de la SLA
L’hétérogénéité de la sclérose latérale amyotrophique (SLA) rend difficile l’évaluation de l’évolution de la maladie. Des échelles telles que l’ALSFRS-R peuvent fournir une orientation, mais elles atteignent aussi leurs limites en situation réelle, comme le montre une étude récente.
La SLA est une maladie neurodégénérative rare qui se caractérise par la perte progressive des neurones moteurs entraînant une faiblesse musculaire, une paralysie et, à terme, le décès.1,2Les patient·es atteint·es de SLA présentent une multitude de symptômes qui limitent successivement les activités de la vie quotidienne (AVQ) et impactent la qualité de vie.3,4L’évolution de la maladie peut être divisée en étapes cliniques et fonctionnelles, qui sont évaluées dans le cadre clinique à l’aide de l’échelle ALSFRS-R (ALS Functional Rating Scale – Revised).5–7La sensibilité de cette échelle semble cependant être insuffisante, en particulier aux stades précoces et avancés de la maladie. De plus, le profil clinique de chaque patient·e n’est pas toujours bien représenté, car quatre domaines fonctionnels différents et plus ou moins impactés (fonction bulbaire, motricité fine et globale, et fonction respiratoire) sont regroupés.8–11
Étude du contexte réel portant sur la limitation des AVQ liée à l’évolution de la maladie
L’étude présentée ici a analysé les données du programme spécifique à la SLA (Adelphi), qui comprenait des questionnaires sur la charge de travail des neurologues (n=59), leur point de vue sur le traitement de la SLA et des formulaires de patient·es (n=379). Des informations sur 24 AVQ différentes tout au long de l’évolution de la maladie étaient également incluses.12–15 L’objectif était d’obtenir un aperçu de l’interprétation du score ALSFRS-R dans le cadre du suivi de l’évolution de la maladie et de la perte fonctionnelle dans la SLA.
Échelle ALSFRS-R à la lumière des données en situation réelle
Les données probantes en situation réelle ont pu mettre en évidence qu’un certain nombre d’AVQ, dont le ménage, le jardinage, la marche sans assistance, la cuisine, les courses et les voyages, étaient déjà fortement associées à une perte d’autonomie à un score ALSFRS-R plus élevé (c’est-à-dire à un stade moins avancé de la maladie) par rapport à d’autres AVQ. Chaque baisse d’un point sur l’échelle ALSFRS-R était associée à la perte d’un niveau d’autonomie, ce qui montre un lien clair entre le score ALSFRS-R et le niveau d’autonomie dans les AVQ. Cependant, les modifications observées sur l’échelle ALSFRS-R ne pouvaient pas refléter entièrement l’impact de l’évolution de la maladie sur les AVQ. D’autres facteurs qui ne sont pas pris en compte ici jouent également un rôle décisif. L’étape la plus avancée de la maladie, à savoir la mise en place d’une sonde d’alimentation, était associée à un score ALSFRS-R moyen de 25,1. Lorsque le score ALSFRS-R atteint 24 points, il faut donc s’attendre à un besoin d’assistance plus important (Fig.1). Cela signifie aussi que l’échelle ALSFRS-R perd de sa pertinence aux stades avancés, car l’autonomie des patient·es peut déjà être complètement perdue ou considérablement réduite à un score ≤24.16
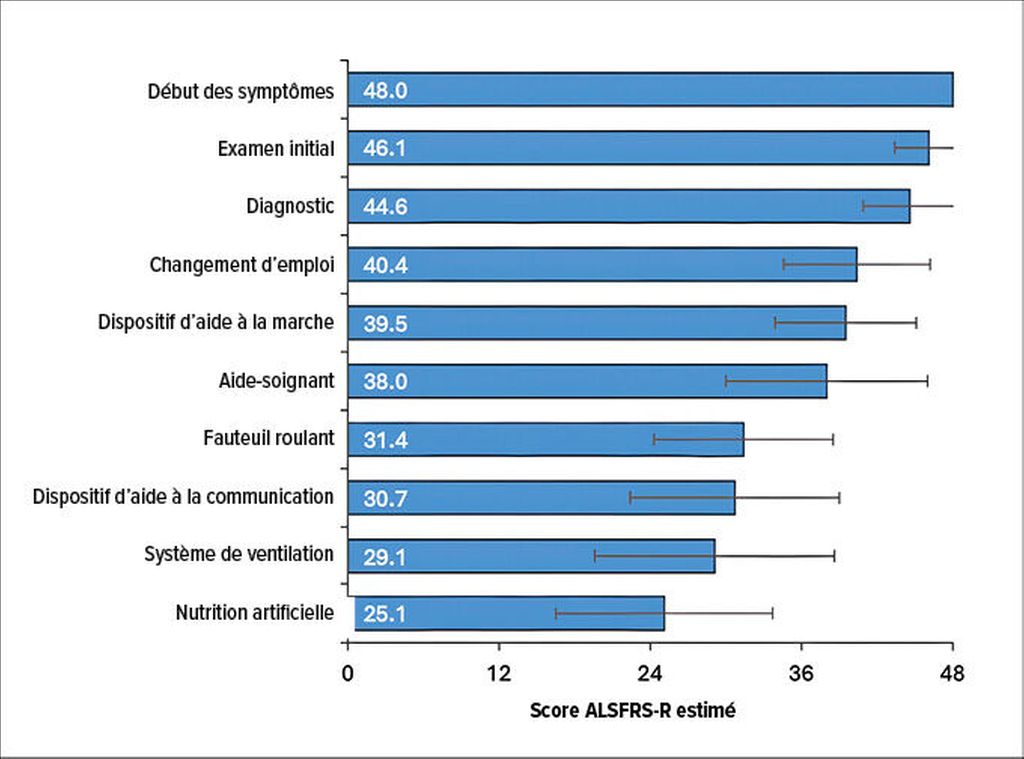
Fig.1: Scores ALSFRS-R à des étapes sélectionnées de la maladie (modifiée selon Mehdiyoun NF et al.)16
Approches pour l’extension de l’échelle ALSFRS-R
Ensemble, les résultats de l’étude ont mis en évidence les limites de l’échelle ALSFRS-R: la pondération homogène des quatre domaines fonctionnels ne semble pas adaptée à la représentation de l’évolution hétérogène de la SLA, en particulier pour l’évaluation des patient·es individuel·les. Il est donc nécessaire de compléter l’échelle ALSFRS-R par des points dans les différents domaines fonctionnels et/ou d’inclure d’autres mesures afin de permettre une évaluation complète de l’état des patient·es.
Source:
Mehdiyoun NF et al.: Evaluating ALSFRS-R as an indicator of disease milestones and functional independence: An observational study of US neurologists and their patients with amyotrophic lateral sclerosis. J Neurol Sci. 2026; 481: 125732
Littérature:
1 Bottero V et al.: Key disease mechanisms linked to amyotrophic lateral sclerosis in spinal cord motor neurons. Front Mol Neurosci 2022; 15: 825031 2 Su WM et al.: Predictors of survival in patients with amyotrophic lateral sclerosis: a large meta-analysis. EBioMedicine. 2021; 74: 103732 3 Zizzi C et al.: Patient reported impact of symptoms in amyotrophic lateral sclerosis (PRISM-ALS): a national, cross-sectional study. EClinicalMedicine 2022; 55: 101768 4 Caballero-Eraso C et al.: Prospective study to evaluate quality of life in amyotrophic lateral sclerosis. Sci Rep 2023; 13(1): 12074 5 Cedarbaum JM et al.: The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999; 169(1-2): 13-21 6 Kollewe K et al.: ALSFRS-R score and its ratio: a useful predictor for ALS-progression. J Neurol Sci 2008; 275(1-2): 69-73 7 Gebrehiwet P et al.: Time from amyotrophic lateral sclerosis symptom onset to key disease milestones: analysis of data from a multinational cross-sectional survey. Amyotroph Lateral Scler Frontotemporal Degener 2024; 25(3-4): 345-57 8 Mandrioli J et al.: Heterogeneity in ALSFRS-R decline and survival: a population-based study in Italy. Neurol Sci 2015; 36(12): 2243-52 9 He R et al.: Milano-torino staging and long-term survival in chinese patients with amyotrophic lateral sclerosis. Cells 2021; 10(5): 1220 10 Wicks P et al.: Measuring function in advanced ALS: validation of ALSFRS-EX extension items. Eur J Neurol 2009; 16(3): 353-9 11 Young CA et al.: Improving the measurement properties of the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R): deriving a valid measurement total for the calculation of change. Amyotroph Lateral Scler Frontotemporal Degener 2024; 25(3-4): 400-9 12 Anderson P et al.: Real-world physician and patient behaviour across countries: Disease-specific programmes - a means to understand. Curr Med Res Opin 2008; 24(11): 3063-72 13 Anderson P et al.: Real-world evidence generation from patients, their caregivers and physicians supporting clinical, regulatory and guideline decisions: an update on disease specific programmes. Curr Med Res Opin 2023; 39(12): 1707-15 14 Babineaux SM et al.: Evidence for validity of a national physician and patient-reported, cross-sectional survey in China and UK: the disease specific programme. BMJ Open 2016; 6(8): e010352 15 Higgins V et al.: Trends in medication use in patients with type 2 diabetes mellitus: a long-term view of real-world treatment between 2000 and 2015. Diabetes Metab Syndr Obes 2016; 9: 371-80 16 Mehdiyoun NF et al.: Evaluating ALSFRS-R as an indicator of disease milestones and functional independence: an observational study of US neurologists and their patients with amyotrophic lateral sclerosis. J Neurol Sci 2026; 481: 125732
Das könnte Sie auch interessieren:

Nouvelles options thérapeutiques pour les troubles psychiatriques
Autour du thème «Santé mentale: améliorer la prise en charge et élargir les horizons», des expert·es de renom en psychiatrie se sont réuni·es à Prague à l’occasion du 34e Congrès ...

Trouble de l’insomnie – du symptôme au diagnostic et au traitement fondé sur les preuves
L’insomnie compte parmi les problèmes de santé les plus fréquents dans la pratique clinique quotidienne. Des travaux récents montrent que le trouble de l’insomnie chronique ne se ...

Focus sur les prophylaxies alternatives et séquentielles de la migraine
Pour les patient·es migraineux·euses ne répondant pas aux anticorps monoclonaux dirigés contre le «calcitonin gene-related peptide» (anti-CGRP-mAbs), des traitements alternatifs et des ...