Plus qu’un problème de sécheresse: gros plan sur la maladie de Sjögren
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
La maladie de Sjögren est une maladie auto-immune systémique inflammatoire chronique présentant une grande variabilité clinique. Elle fait partie de la famille des collagénoses, à laquelle appartiennent notamment la sclérose systémique, le lupus érythémateux disséminé ou la polymyosite. Cet article de synthèse donne un aperçu actuel du tableau clinique, du diagnostic et du traitement, et vise à sensibiliser les médecins internistes à la prise en charge différenciée de cette maladie souvent sous-estimée.
La maladie de Sjögren (SjD) se caractérise par une infiltration lymphocytaire des glandes exocrines, notamment des glandes salivaires et lacrymales, ce qui entraîne un dysfonctionnement. Il en résulte les symptômes buccaux ou oculaires typiques du syndrome sec. La sécheresse peut aussi toucher le nez, la gorge, la peau ou la vulve/le vagin. En outre, environ un tiers des personnes atteintes présentent une atteinte extra-glandulaire, pratiquement tous les systèmes d’organes pouvant être concernés.
Terminologie
Le nom de la maladie vient de l’ophtalmologue suédois Henrik Sjögren qui, dans sa thèse publiée en allemand en 1933, a décrit une kératoconjonctivite sèche chez 19 femmes, dont 13 présentaient également des manifestations articulaires.1 Au niveau international, le terme «maladie de Sjögren» (en anglais «Sjögren’s disease») s’est imposé ces dernières années sur recommandation des expert·es et des personnes concernées par rapport à l’ancien terme «syndrome de Sjögren». Cela permet de tenir compte du fait qu’il s’agit d’une maladie systémique qui doit être prise au sérieux et non d’une succession de symptômes, comme le terme «syndrome» pourrait le suggérer.
La SjD peut exister en tant que maladie isolée («maladie de Sjögren primaire») ou être associée à une autre maladie auto-immune inflammatoire chronique, telle que la polyarthrite rhumatoïde ou le lupus érythémateux disséminé. Dans le second cas, ce que l’on appelait autrefois «syndrome de Sjögren secondaire» est aujourd’hui dénommé «maladie de Sjögren associée».
Pathogenèse et étiologie
La pathogenèse de la SjD est complexe et n’est pas complètement élucidée. Les prédispositions génétiques, les facteurs environnementaux ainsi qu’hormonaux jouent un rôle central.
Les facteurs environnementaux supposés sont notamment les infections par le virus d’Epstein-Barr (EBV) et le cytomégalovirus (CMV).2 De plus, l’hypoœstrogénie associée à la ménopause semble déclencher le développement de la maladie, ce qui pourrait expliquer le fait qu’elle soit souvent diagnostiquée chez les femmes ménopausées.3 Dans le cas de la SjD, le processus auto-immun se manifeste principalement par l’atteinte des glandes exocrines, la formation d’auto-anticorps, le dépôt de complexes immuns ainsi que par l’infiltration lymphocytaire de différents organes dans le cadre d’une évolution systémique. Une production accrue du facteur d’activation des cellules B (BAFF) ainsi qu’une activation excessive des cellules B, due à une modification de l’expression du récepteur BAFF dans différents types de cellules immunitaires, jouent un rôle central.4
Tableau clinique
Le symptôme principal de la SjD est le syndrome sec. La sécheresse oculaire peut se manifester par des brûlures et des démangeaisons oculaires, une sensation de sable et la nécessité d’utiliser un collyre hydratant. Certaines personnes concernées ne ressentent aucune sécheresse oculaire, bien que celle-ci soit détectable par des mesures quantitatives et qualitatives.
La sécheresse buccale est souvent accentuée la nuit, peut entraîner une dysgueusie et une dysphagie, notamment des aliments secs comme le pain. Les conséquences du manque de salive sont, entre autres, un risque accru de caries et de muguet buccal.5
Chez certaines personnes atteintes, on observe des gonflements récurrents des glandes salivaires, souvent autolimités.
De nombreuses personnes se plaignent également de myalgies et d’arthralgies, qui ne sont souvent pas cliniquement détectables comme une myosite ou une arthrite manifeste et qui ressemblent parfois au tableau clinique de la fibromyalgie.6
Jusqu’à 70% des personnes atteintes de SjD souffrent également de fatigue chronique, bien qu’un lien direct avec les voies de signalisation pro-inflammatoires n’ait pas pu être établi.7
Les manifestations extra-glandulaires sont multiples. Il n’est pas rare que la SjD soit uniquement diagnostiquée dans le cadre du bilan étiologique d’une polyneuropathie, d’une pneumopathie interstitielle diffuse, d’une néphrite tubulo-interstitielle ou d’une glomérulonéphrite indéterminées. Dans de rares cas, les auto-anticorps anti-SSA/Ro typiques de la SjD peuvent entraîner un bloc AV congénital à partir de la 16e semaine de grossesse. Celui-ci peut également être à l’origine d’examens approfondis, au cours desquels le diagnostic d’une SjD est finalement posé.
En outre, la SjD peut entraîner de l’arthrite, typiquement non érosive, qui touche souvent de manière symétrique les articulations métacarpo-phalangiennes, interphalangiennes proximales et les poignets. L’atteinte cutanée va des manifestations de type vascularite telles que les pétéchies et le purpura à l’érythème annulaire ou aux lésions de type érythème polymorphe. Certaines personnes présentent un phénomène de Raynaud déclenché par le froid, qui est souvent léger et qui, contrairement à la sclérose systémique, n’entraîne typiquement pas de troubles trophiques tels que des ulcères ou des «pitting scars». La microscopie capillaire révèle généralement un résultat normal ou des modifications non spécifiques telles qu’une diminution de la densité capillaire, mais aucune microangiopathie organique.8
La Figure 1 fournit un aperçu des manifestations systémiques les plus fréquentes dans la SjD.
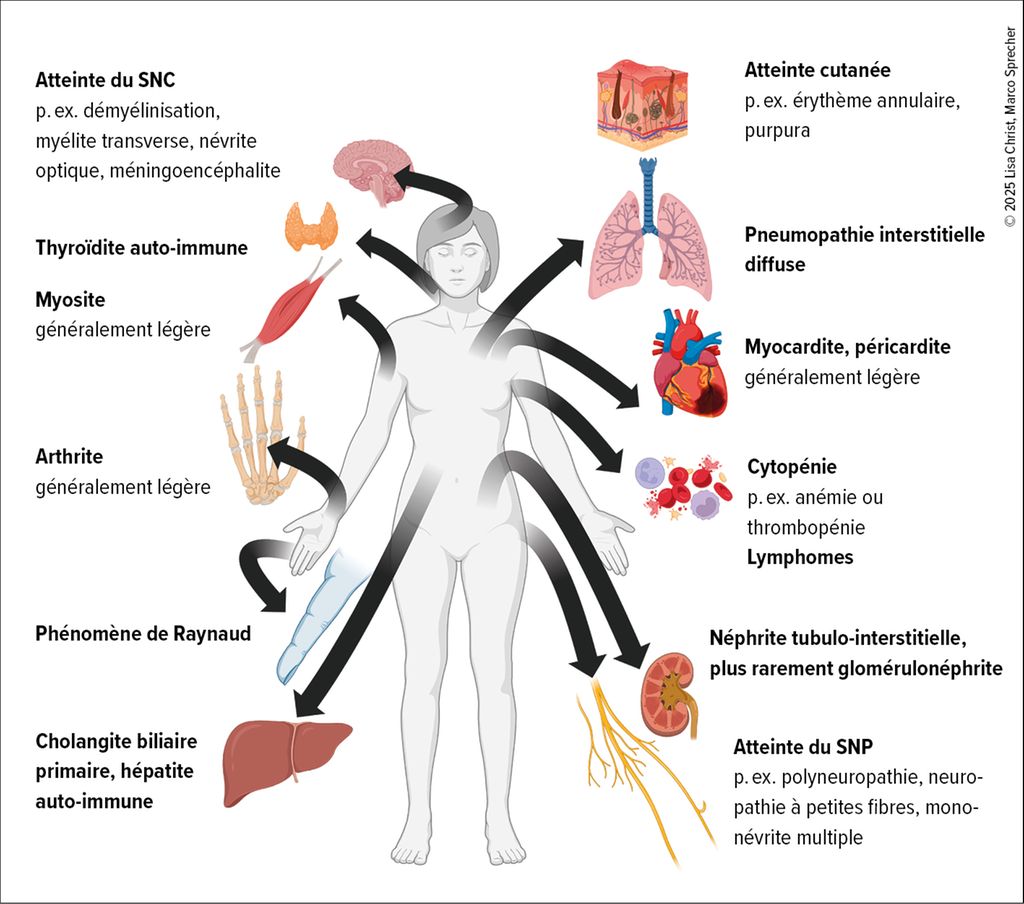
Fig.1: Aperçu des manifestations extra-glandulaires les plus fréquentes de la maladie de Sjögren (graphique réalisé avec © Biorender)
Diagnostic
Le diagnostic repose sur une combinaison d’évaluation clinique, de résultats d’analyses de laboratoire et de tests fonctionnels. Les critères de classification actuellement en vigueur de l’EULAR et de l’American College of Rheumatology (ACR) comprennent cinq domaines, auxquels sont attribués un nombre de points différent (Tab.1). Un score ≥4 points permet la classification comme SjD.
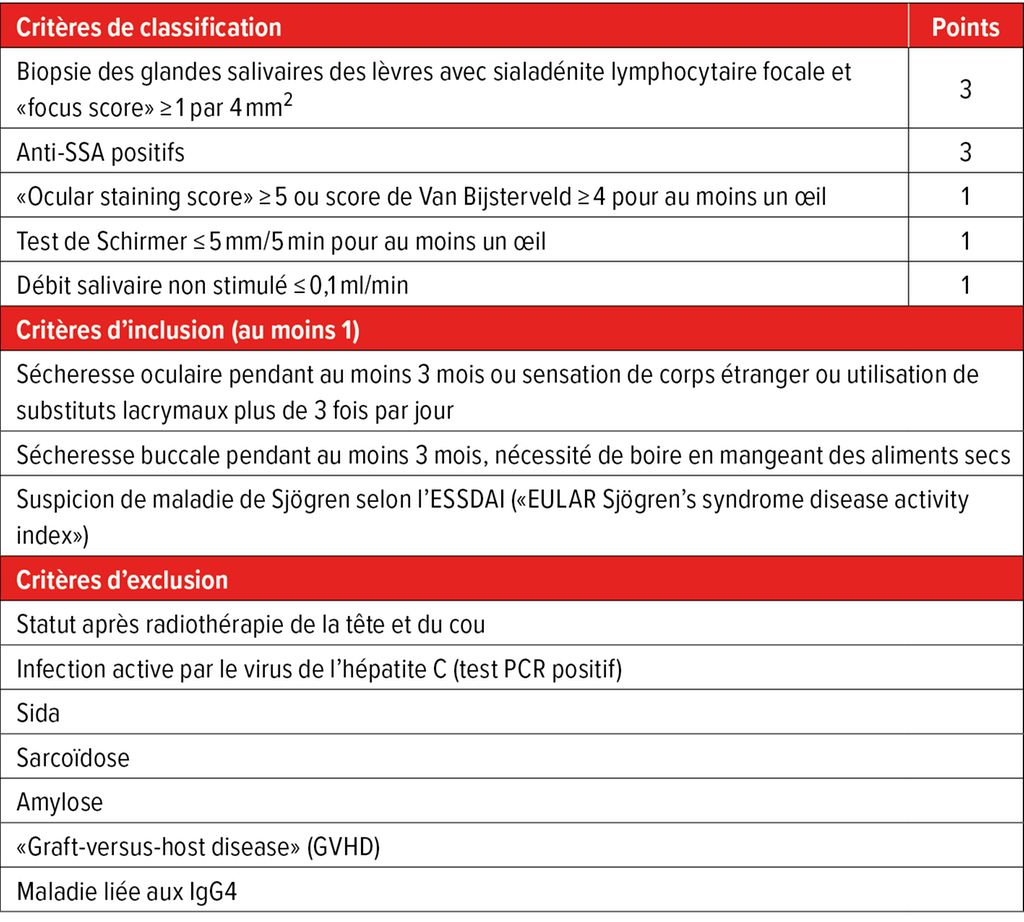
Tab.1: Critères de classification 2016 de l’ACR/EULAR pour la maladie de Sjögren. Les critères de classification sont remplis si au moins 4 points, au moins 1 critère d’inclusion et 0 critère d’exclusion sont atteints
Les marqueurs sérologiques typiques sont les anticorps antinucléaires (AAN), les anticorps anti-Ro/SSA et anti-La/SSB, les facteurs rhumatoïdes ainsi qu’une hypergammaglobulinémie polyclonale, et dans certains cas, des cryoglobulines. Environ 60% des personnes atteintes de SjD présentent des facteurs rhumatoïdes positifs, ce qui reflète une plus grande activité des cellules B et va souvent de pair avec une plus grande activité systémique de la maladie.9 Environ 15% des personnes atteintes de SjD sont négatives pour les ANA, les anticorps anti-Ro/SSA, les anticorps anti-La/SSB et les facteurs rhumatoïdes.10
Les tests fonctionnels tels que le test de Schirmer et le débit salivaire sont des paramètres importants pour objectiver les symptômes du syndrome sec. Le test de Schirmer consiste à accrocher une bande de papier filtre standardisée dans la paupière inférieure et à mesurer, après cinq minutes, la longueur de la partie du papier humidifiée par le liquide lacrymal. Une valeur de 5mm ou moins est considérée comme pathologique. Lors du débit salivaire non stimulé, la salive est recueillie en position assise, sans stimuli extérieurs, pendant une période définie (généralement 5 à 15 minutes) en la collectant librement dans un récipient de mesure. Le débit salivaire stimulé est mesuré après un stimulus contrôlé, par exemple en mâchant de la paraffine standardisée ou un chewing-gum, la salive produite étant également recueillie et mesurée dans un laps de temps déterminé. Souvent, un débit salivaire non stimulé ≤0,1ml/min est considéré comme pathologique, ce qui est toutefois controversé, car cette valeur ne tient pas compte des variations physiologiques spécifiques à l’âge et au sexe, et a une sensibilité très faible (environ 50%), contrairement au test de Schirmer (environ 70%). Certain·es expert·es recommandent donc une valeur seuil ≤0,2ml/min.11

Fig. 2: Échographie de la glande parotide, grade 2 selon l’OMERACT: manques d’uniformité de la texture de la glande parotide avec de multiples zones hypo- à anéchogènes qui sont encore entourées de tissu normal
Les techniques d’imagerie telles que l’échographie des glandes parotide et submandibulaire ont pris de l’importance au cours des dernières années, car elles sont sensibles, non invasives et rapides à réaliser. Sur l’image B, une sonde linéaire de 10 à 18 MHz permet de constater des manques d’uniformité plus ou moins marqués du parenchyme des glandes salivaires, avec des zones arrondies focales hypo- ou anéchogènes (Fig.2). La classification de ces modifications en différents grades (0–3) par l’OMERACT («Outcome Measures in Rheumatology Clinical Trials») a été largement acceptée au niveau international.12 L’échographie compare les glandes salivaires au parenchyme de la thyroïde, qui doivent normalement être homogènes et isoéchogènes entre elles. L’hypervascularisation peut également être quantifiée par doppler couleur et classée en différents grades (0–3) selon l’OMERACT.13 Il convient de s’abstenir de manger et de mâcher au moins une heure avant, car le processus de mastication stimule physiologiquement la circulation sanguine.
Un article de synthèse systématique avec méta-analyse a montré que l’échographie des glandes salivaires présente une sensibilité groupée de 80% (IC à 95%: 77–83%) ainsi qu’une spécificité de 90% (IC à 95%: 87–92%) pour le diagnostic.14 Une autre étude a en outre constaté une forte concordance des résultats de l’échographie avec les résultats de la biopsie de la glande parotide (83%) ou de la lèvre inférieure (79%). Dans leur étude, un résultat d’échographie pathologique combiné avec la détection d’auto-anticorps anti-Ro/SSA ou une échographie normale combiné avec l’absence de ces anticorps était particulièrement significative: les deux constellations présentaient une valeur prédictive élevée pour le diagnostic de la maladie de Sjögren ou son exclusion.15
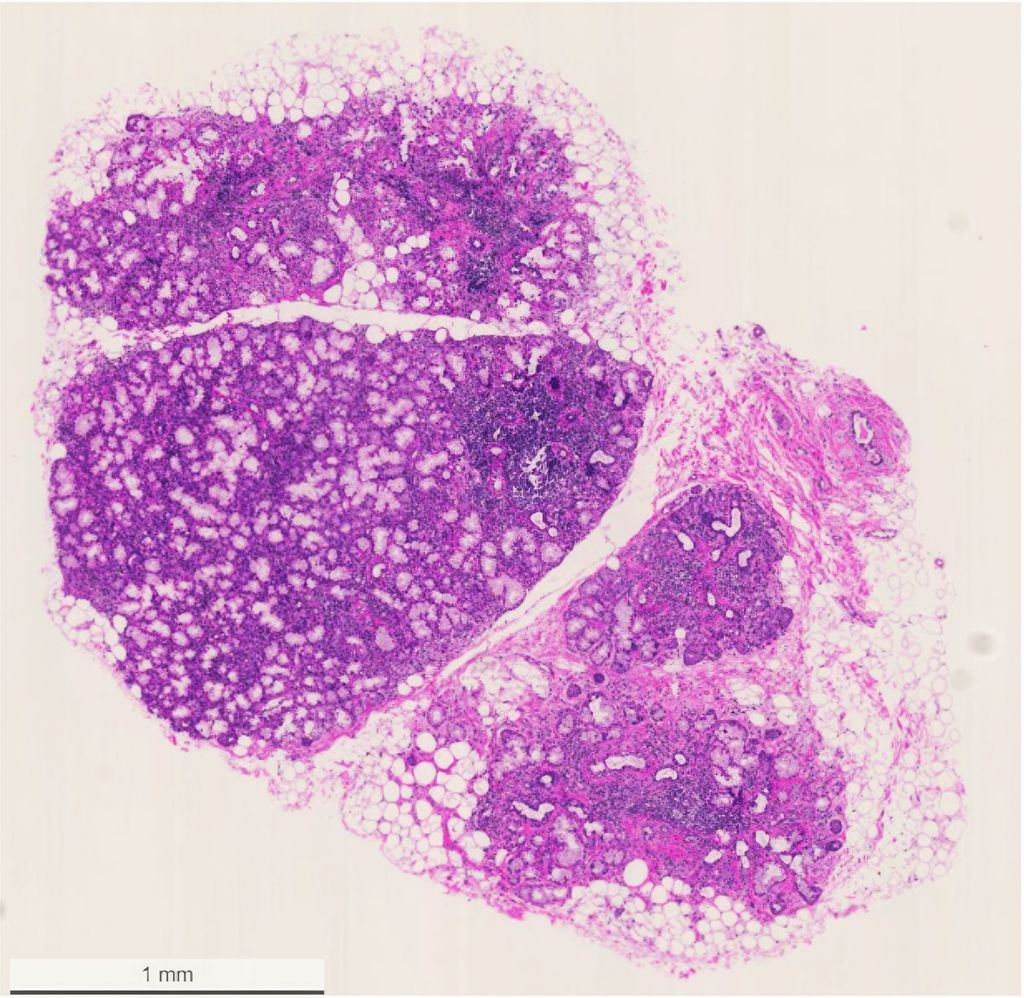
Fig. 3: Biopsie des glandes salivaires au niveau de la lèvre inférieure avec mise en évidence de multiples foyers lymphocytaires (avec l’aimable autorisation de la patiente)
La biopsie des glandes salivaires reste néanmoins un élément central du diagnostic, mais elle n’est pas obligatoire si le reste du tableau clinique est clair. Sous anesthésie locale, plusieurs lobes de glandes salivaires sont prélevés, en général sur la face interne de la lèvre inférieure, en prenant soin de ne pas endommager les vaisseaux et les nerfs. La mise en évidence histologique de foyers lymphocytaires – infiltrats focaux, généralement périduraux, composés d’au moins 50 lymphocytes et histiocytes – est caractéristique de la SjD (Fig.3). Le «focus score» décrit le nombre de ces foyers par 4mm2 de tissu glandulaire. Un score ≥1 est considéré comme déterminant pour le diagnostic de la SjD, mais n’est pas pathognomonique. Par exemple, une infection par le VIH non traitée peut avoir une présentation histologique très similaire dans les glandes salivaires.16 D’autre part, la biopsie des glandes salivaires contribue fortement au diagnostic différentiel, par exemple pour faire la distinction avec les maladies liées aux IgG4, la sarcoïdose, l’amylose ou les processus malins, qui peuvent également être à l’origine des symptômes du syndrome sec. La mise en évidence de centres germinaux ectopiques est en outre importante pour le pronostic, car le risque de lymphome peut être multiplié par 7,8, selon les études.17
Risque de lymphome
Il convient de porter une attention particulière au risque accru de développer un lymphome non hodgkinien à cellules B, qui survient chez environ 5 à 10% des personnes atteintes de SjD.18,19 Parmi les facteurs de risque, on retrouve notamment un gonflement persistant de la glande parotide, une hypocomplémentémie, une cryoglobulinémie et une gammapathie monoclonale.20 En revanche, les personnes atteintes de SjD séronégatives présentent un risque de lymphome plus faible, de l’ordre de 1% selon les études,18 mais toujours supérieur à celui de la population générale.
Sur le plan histologique, il s’agit le plus souvent de lymphomes de la zone marginale extranodale (lymphomes MALT), qui se manifestent généralement dans les glandes salivaires ou d’autres tissus muqueux. Sur le plan clinique, ils peuvent se caractériser par une lymphadénopathie, une organomégalie ou des symptômes systémiques (symptômes B). Le suivi des personnes à risque passe par des examens cliniques réguliers, des paramètres d’évolution sérologiques (p.ex. numération formule sanguine, complément, bêta-2-microglobuline, rapport CD4/8) et, le cas échéant, par l’imagerie médicale. Il n’existe pas à ce jour de consensus international sur le suivi précis. Un diagnostic précoce et une prise en charge interdisciplinaire sont toutefois déterminants pour un bon pronostic.
Traitement
Le traitement de la SjD s’oriente en fonction des symptômes, de la sévérité de la maladie ainsi que du type et du degré d’atteinte des organes. Il existe certes des recommandations thérapeutiques comme celles de l’European League Against Rheumatism (EULAR)21, mais il n’existe à ce jour aucun schéma thérapeutique uniforme basé sur des preuves, raison pour laquelle une approche individualisée et multidisciplinaire est recommandée.
Traitement des symptômes du syndrome sec
Le traitement symptomatique du syndrome sec constitue la référence et doit être adapté individuellement ainsi qu’évalué régulièrement.
Symptômes oculaires du syndrome sec
Les substituts lacrymaux constituent le premier choix. Il existe à cet effet sur le marché des collyres contenant divers principes actifs, par exemple l’acide hyaluronique, le carbomère, l’hypromellose, la povidone K25 ou la carmellose. Si des applications très fréquentes par jour sont nécessaires, il convient de privilégier des préparations sans conservateurs, car ces derniers peuvent irriter davantage les yeux en cas d’utilisation fréquente. Les gels ophtalmiques visqueux ou les pommades ophtalmiques contenant de la vitamine A sont particulièrement recommandés pour la nuit, car ils permettent une hydratation prolongée de la surface oculaire, mais peuvent entraîner une vision floue temporaire en raison de leur consistance. Les préparations contenant des lipides ralentissent l’altération ou l’évaporation du film lacrymal. Elles compensent ainsi avant tout un dysfonctionnement des glandes de Meibomius, qui explique également une partie des symptômes oculaires du syndrome sec dans la SjD.22
Pour prolonger la durée d’efficacité du film lacrymal et réduire le larmoiement, il est possible d’insérer des «punctual plugs» dans les canalicules lacrymaux, de manière temporaire ou permanente selon l’évolution. Dans les cas sévères et réfractaires, on peut en outre utiliser un collyre à base de sérum autologue. En cas de kératoconjonctivite sèche modérée à sévère, il est également possible de recourir à une immunosuppression topique à l’aide de collyres à base de ciclosporine.
En complément, des mesures non médicamenteuses telles que des soins réguliers du bord des paupières, le port de lunettes à chambre humide («moisture chamber glasses») ainsi qu’une humidification de l’air à l’intérieur peuvent s’avérer utiles pour réduire l’évaporation du film lacrymal et atténuer les symptômes.
Symptômes buccaux du syndrome sec
Le traitement de la sécheresse buccale comprend à la fois des mesures de substitution et de stimulation des sécrétions. Les substituts salivaires tels que les sprays buccaux, les comprimés à sucer ou les gels hydratants, par exemple à base de carmellose ou de xylitol, peuvent soulager les symptômes subjectifs et protéger les muqueuses. En complément, il est recommandé de mâcher du chewing-gum sans sucre ou de consommer des bonbons acides sans sucre afin de stimuler la sécrétion salivaire de manière physiologique.
Les personnes atteintes de SjD dont la fonction résiduelle des glandes salivaires est préservée peuvent tirer profit de la pilocarpine (5mg, 3 à 4 fois par jour) ou de la céviméline (30mg, 3 fois par jour, le cas échéant) pour stimuler la sécrétion. Ces substances sont toutefois associées à des effets secondaires potentiels tels qu’une transpiration accrue, des bouffées vasomotrices ou des diarrhées, c’est pourquoi leur utilisation doit être évaluée au cas par cas.
L’accent est également mis sur la prévention des complications dentaires: une hygiène buccale minutieuse avec utilisation d’un dentifrice fluoré, des contrôles dentaires réguliers et l’évitement des aliments cariogènes sont essentiels. En raison de la modification de la microflore buccale et de l’affaiblissement des défenses immunitaires, il faut en outre tenir compte d’une sensibilité accrue aux infections à Candida. Des antifongiques à action locale peuvent être utilisés dans ce cas.
Traitement systémique
En cas de manifestations extra-glandulaires, un traitement systémique immunomodulateur ou immunosuppresseur peut être indiqué. Le choix des médicaments dépend de l’atteinte des organes, de l’activité de la maladie et des comorbidités individuelles.
L’hydroxychloroquine constitue un traitement souvent bien toléré, notamment en cas d’arthralgie, de manifestations cutanées, ainsi que pour réduire le risque de bloc de conduction congénital en cas d’auto-anticorps anti-Ro/SSA positifs. Des contrôles ophtalmologiques réguliers sont nécessaires, car la rétinopathie potentiellement induite par l’hydroxychloroquine est une complication rare, mais grave, généralement réversible si le diagnostic est posé à temps.
En cas de maladie systémique active, les glucocorticoïdes sont souvent utilisés. Ils peuvent être administrés initialement à des doses élevées (p.ex. prednisone: 0,5–1mg/kgPC/j) et doivent ensuite être rapidement réduits à une dose d’entretien aussi faible que possible afin d’éviter les effets secondaires à long terme.
Les antirhumatismaux modificateurs de la maladie (ARMM) conventionnels tels que le méthotrexate, le léflunomide, l’azathioprine ou le mycophénolate mofétil peuvent être pertinents en fonction des manifestations organiques systémiques. Leur action est souvent retardée, c’est pourquoi il est courant de les associer à des glucocorticoïdes au début.
En cas d’évolution sévère et réfractaire ou dans certaines constellations à risque (p.ex. polyneuropathie avec vascularite, atteinte du SNC, suspicion de lymphome), on peut envisager l’utilisation de produits biologiques, en recourant souvent à un traitement de déplétion des cellules B par le rituximab ou à une inhibition du BAFF par le belimumab. Il s’agit d’une utilisation «off-label» qui a généralement lieu dans des centres spécialisés.
Perspectives
La recherche sur la SjD s’est nettement accélérée au cours des dernières années. Plusieurs études de phase II ont atteint pour la première fois leurs critères d’évaluation primaire: un signal clair pour les progrès thérapeutiques futurs.
À titre d’exemple, l’anticorps anti-récepteur BAFF ianalumab a permis d’obtenir une réduction significative de l’activité de la maladie (ESSDAI) et une amélioration du débit salivaire dans une vaste étude de phase II randomisée, contrôlée par placebo.23 L’anticorps anti-CD40 iscalimab, qui intervient de manière ciblée dans l’interaction entre les cellules T et B, a également montré une efficacité clinique pertinente sur l’activité de la maladie (ESSDAI) dans une étude de preuve de concept (PoC).24
D’autres substances comme la protéine de fusion RNase-Fc RSLV-132 ou la «small molecule» iguratimod font actuellement l’objet d’études de phase IIb. Leurs mécanismes d’action complexes ouvrent la voie à de nouvelles options thérapeutiques allant de la liaison ciblée d’auto-anticorps à l’inhibition des voies de signalisation pro-inflammatoires.
Dans l’ensemble, on constate une tendance aux approches stratifiées, qui s’adressent de manière plus différenciée aux sous-groupes immunopathologiques. Si l’efficacité de ces approches thérapeutiques innovantes est confirmée dans de vastes études de phase III, le traitement de la SjD est sur le point de connaître un changement de paradigme.
Littérature:
1Sjögren H: Acta Ophthalmol 1933; 2: 1-151 2 Kivity S et al.: J Autoimmun 2014; 51: 17-22 3 Punnanitinont A, Kramer JM: Front Dent Med 2023; 4: 1168645 4 Thompson N et al.: Rheumatology 2016; 55: 1548-55 5 Napeñas JJ, Rouleau Oral Maxillofac Surg Clin North Am 2014; 26: 55-62 6 Gau SY et al.: Front Immunol 2021; 12: 640618 7 Mæland E et al.: Front Immunol 2021; 12: 703079 8 Melsens K et al.: Clin Exp Rheumatol 2020; 38(Suppl 126): 150-7 9 Maślińska M et al.: Clin Rheumatol 2019; 38: 1301-7 10 Yazisiz V et al.: Clin Rheumatol 2021; 40: 221-9 11 Lacombe V et al.: Arthritis Res Ther 2020; 22: 38 12 Finzel S et al.: Rheumatology 2021; 60: 2169-76 13 Hočevar A et al.: Rheumatology 2022; 61: 3341-50 14 Ramsubeik K et al.: Ther Adv Musculoskelet Dis 2020; 12: 1759720X20973560 15 Mossel E et al.: Ann Rheum Dis 2017; 76: 1883-9 16 Kordossis T et al.: Br J Rheumatol 1998; 37: 691-5 17 Sène D et al.: Arthritis Rheumatol 2018; 70: 1481-8 18 Quartuccio L et al.: Autoimmun Rev. 2015; 14: 1019-22 19 Voulgarelis M et al.: Medicine 2012; 91: 1-9 20 Retamozo S et al.: Lupus 2019; 28(8): 923-36 21 Ramos-Casals M et al.: Ann Rheum Dis 2020; 79: 3-18 22 Noh SR et al.: Int Ophthalmol 2022; 42: 191-200 23 Bowman SJ et al.: Lancet 2022; 399: 161-71 24 Fisher B et al.: Lancet Rheumatol 2020; 2: e142-52
Das könnte Sie auch interessieren:
Traitement hypolipémiant chez les personnes vivant avec le VIH
Les personnes vivant avec le virus de l’immunodéficience humaine (VIH) présentent un risque accru de maladies cardiovasculaires athéroscléreuses. De plus, le traitement antirétroviral et ...
Le Pelargonium sidoides est-il une option thérapeutique chez les enfants atteints du syndrome mains-pieds-bouche?
Une étude a examiné pour la première fois l’extrait de racine de Pelargonium sidoides EPs®7630 chez des enfants atteints du syndrome mains-pieds-bouche et a démontré une réduction ...

Prise en charge des acouphènes en 2025
Le traitement des acouphènes a évolué au cours des dernières années. Les directives de l’AWMF sur la prise en charge des acouphènes proposent des recommandations fondées sur des preuves ...