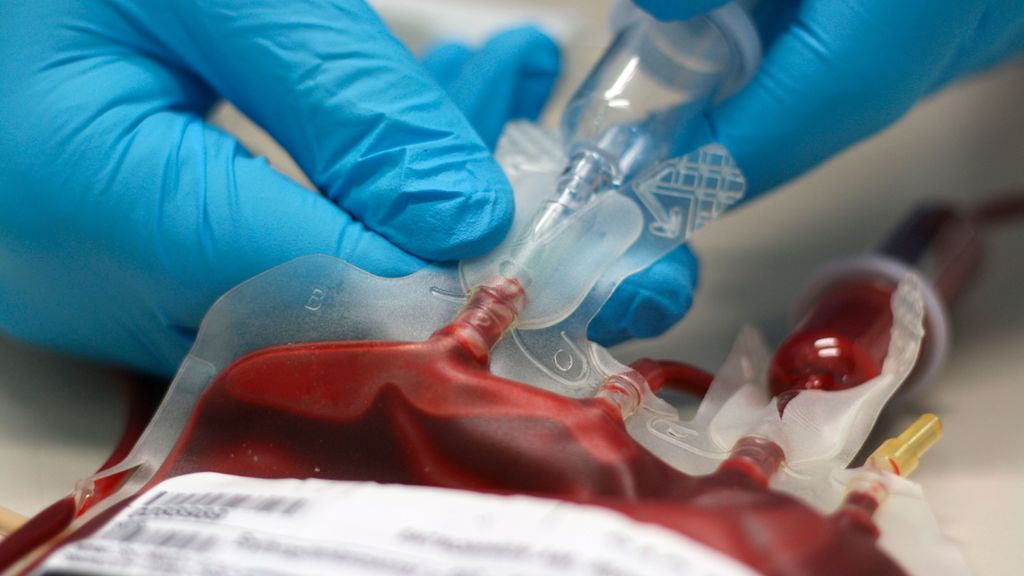
Mise à jour: comment éviter les transfusions?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Chez les patient·es à faible risqueatteint·es de syndromes myélodysplasiques (SMD), l’anémie nécessitant des transfusions constitue souvent le principal problème altérant la qualité de vie. De nouvelles approches thérapeutiques dans ce domaine apportent d’ores et déjà des progrès significatifs et laissent espérer de nouvelles améliorations à l’avenir.
Keypoints
-
L’anémie nécessitant des transfusions est la principale caractéristique des SMD à faible risque, avec des conséquences sur la qualité de vie, la morbidité et la mortalité.
-
L’EPO, les transfusions, la chélation du fer ainsi que, dans des situations spécifiques, le lénalidomide et le traitement immunosuppresseur constituent des options thérapeutiques éprouvées de longue date.
-
Dans une comparaison randomisée en première ligne, le luspatercept présente des avantages par rapport à l’EPO en termes de taux de réponse, de durée de réponse ainsi que de survie globale.
-
Jusqu’à présent, l’imetelstat a été étudié dans le contexte d’une situation réfractaire ou inadéquate à l’EPO et a montré de bonnes réponses, notamment en cas de charge transfusionnelle élevée.
SMD à faible risque
Les syndromes myélodysplasiques (SMD) sont des maladies clonales acquises des cellules souches hématopoïétiques de la moelle osseuse. Une hématopoïèse inefficace entraîne des cytopénies persistantes associées à un risque accru de progression vers une leucémie myéloïde aiguë.1
Les SMD à faible risque présentent une proportion moindre de blastes médullaires, des cytopénies légères à modérées ainsi que des anomalies cytogénétiques plus favorables, associées à une survie médiane plus longue et à un risque de progression réduit.
Différents scores pronostiques ont été développés afin de définir les groupes à risque. L’IPSS-R est établi de longue date et largement utilisable.2 L’IPSS-M, qui intègre des données moléculaires, classe de nombreux·ses patient·es dans des catégories de risque plus élevées, ce qui nécessite encore une validation plus poussée.3 Pour la dichotomisation en risque faible vs risque élevé, des catégories de scores sont souvent regroupées (p. ex. low+int1 vs int2+high dans l’IPSS, ou very low/low/moderate low vs moderate high/high/very high dans l’IPSS-M). Pour l’IPSS-R, un cut-off peut être fixé à ≥3,5 points.4
L’observation individuelle de l’évolution est utile pour évaluer le risque réel. Cela est particulièrement possible dans les SMD à faible risque, où une stratégie initiale de «watchful waiting» est souvent justifiée.5
Comment traiter les SMD à faible risque?
Le traitement des SMD à faible risque vise principalement à préserver ou améliorer la qualité de vie et à prévenir les complications. Une approche individualisée et graduée est essentielle, allant de l’observation attentive à l’utilisation de traitements médicamenteux ciblés. De nombreux éléments thérapeutiques sont bien établis,5 mais des innovations et des progrès ont été réalisés notamment dans le domaine de l’anémie transfusion-dépendante.
En cas de paramètres hématologiques stables sans complications aiguës, une stratégie de «watchful waiting» est, comme mentionné, justifiée. Les «best supportive care» restent une réalité pour de nombreux·ses patient·es. L’objectif des approches thérapeutiques modernes est de réduire la dépendance aux mesures de soutien, voire d’éviter qu’elles ne deviennent nécessaires.
Dans de rares cas de SMD à faible risque, une thrombocytopénie isolée est au premier plan: une option thérapeutique «off-label» par agonistes du récepteur de la thrombopoïétine (TPO) peut alors être envisagée.6 Dans les SMD hypoplasiques, un traitement par globuline antithymocyte combinée à la ciclosporine est possible – en raison de sa toxicité toutefois uniquement chez des patient·es jeunes et en bon état général.7
La transplantation allogénique de cellules souches constitue une option curative, également réservée à certain·es patient·es jeunes sélectionné·es. Dans les SMD à faible risque, l’indication doit être posée de manière très stricte:8 Une évolution défavorable et rapide ainsi qu’une suspicion de progression peuvent constituer des déclencheurs.
Anémie nécessitant des transfusions dans les SMD à faible risque
Une anémie pouvant nécessiter l’administration régulière de concentrés érythrocytaires est fréquente chez les patient·es atteint·es de SMD à faible risque – au moment du diagnostic déjà, environ 60–66% sont transfusion-dépendant·es. L’indication des transfusions et leur fréquence relèvent de l’activité individuelle de la maladie, des comorbidités et de la symptomatologie clinique.
Les transfusions présentent certains inconvénients potentiels. Chaque administration comporte un risque de complications immunologiques; une allo-immunisation éventuelle peut compliquer les transfusions futures.9 Malgré des standards de sécurité élevés, un risque minimal de transmission d’infections persiste.
La complication la plus fréquente des transfusions chroniques est la surcharge en fer: chaque transfusion apporte du fer supplémentaire dans l’organisme qui, en l’absence de mécanismes d’élimination efficaces, s’accumule dans des organes tels que le foie, le cœur et le pancréas. Cela entraîne à long terme des lésions organiques et nécessite un traitement concomitant par chélation du fer.10
Il est établi depuis longtemps qu’une charge transfusionnelle élevée peut être associée à une réduction de la survie globale.11 Malgré de nombreuses analyses, la part respective des transfusions en tant que telles ou d’une biologie potentiellement plus agressive des SMD transfusion-dépendants reste incertaine. Indépendamment de cela, les traitements visant à éviter les transfusions peuvent avoir un effet bénéfique.
En définitive, il est évident qu’éviter les transfusions tout en obtenant des taux d’hémoglobine suffisants – et plus stables que sous transfusion – peut améliorer la qualité de vie, à condition qu’aucune toxicité supplémentaire liée au traitement médicamenteux alternatif ne survienne.
Comment prévenir les transfusions?
Érythropoïétine
L’érythropoïétine (EPO) est un traitement éprouvé de longue date chez les patient·es atteint·es de SMD à faible risque présentant une anémie symptomatique, en particulier lorsque les taux endogènes d’EPO avant le traitement sont < 500UI/l (idéalement < 200UI/l) et que le besoin transfusionnel est encore absent ou faible («score nordique»).12
La posologie initiale recommandée est de 30000 à 40000UI par semaine par voie sous-cutanée; une augmentation de la dose est possible en cas de réponse insuffisante de l’hémoglobine.
Sous EPO, selon la rigueur d’application du score nordique, jusqu’à 60% des patient·es peuvent obtenir une augmentation significative de l’hémoglobine et une indépendance transfusionnelle; l’administration additionnelle de G-CSF peut encore augmenter les taux de réponse. Une première réponse est généralement observée après 8–12 semaines; la durée médiane de la réponse se situe entre 18 et 24 mois – une réponse pouvant durer plusieurs années est également possible en cas de maintien d’une bonne efficacité. Un contrôle étroit de la réponse devrait conduire à une adaptation individuelle de la dose et, en présence d’alternatives disponibles, à un changement précoce de traitement.
Lénalidomide
Le lénalidomide est autorisé chez les patient·es adultes présentant des SMD à faible risque avec anémie transfusion-dépendante et cytogénétique del(5q) isolée. Dans les études cliniques, environ 67% des patient·es ont obtenu une indépendance transfusionnelle complète, 76% ont réduit leur besoin transfusionnel d’au moins 50% et environ 73% ont présenté une réponse cytogénétique.13 Cette dernière s’accompagne d’une durée médiane d’indépendance transfusionnelle de plus de 80 semaines. Les patient·es répondeur·seuses au lénalidomide bénéficient en outre d’une survie globale significativement prolongée (durée médiane d’OS d’environ 60–80 mois).
Les toxicités dose-limitantes les plus fréquentes sont des neutropénies de grade 3/4 (jusqu’à 60%) et des thrombocytopénies (jusqu’à 50%); les effets indésirables non hématologiques comprennent des éruptions cutanées, une fatigue, des diarrhées ainsi qu’un risque accru de thrombo-embolie veineuse; ils sont généralement contrôlables par des interruptions ou des réductions temporaires de dose.
Chez les patient·es sans del(5q), le lénalidomide permet d’obtenir des taux de réponse érythroïde d’environ 40–45%, avec une indépendance transfusionnelle complète dans environ 15–20% des cas et une durée médiane de réponse d’environ 20–25 semaines.14 Compte tenu du rapport bénéfice-risque, aucune autorisation n’a été obtenue dans cette indication.
Tab.1: Principales options thérapeutiques en cas de SMD à faible risque nécessitant des transfusions
Luspatercept
Le luspatercept stimule l’érythropoïèse en inhibant la signalisation médiée par le TGfβ et a été autorisé en 2023 en Europe pour les patient·es présentant une anémie transfusion-dépendante dans les SMD à faible risque avec sidéroblastes en anneau (RS), insuffisamment répondeur·euses à un traitement à base d’érythropoïétine ou non éligibles à celui-ci (ndlr: non autorisé en Suisse). En 2024, l’autorisation a été étendue indépendamment du statut RS et du statut EPO en tant que traitement de première ligne. L’autorisation repose sur deux études majeures: MEDALIST et COMMANDS.
MEDALIST
Dans l’étude de phaseIII contrôlée par placebo MEDALIST, environ 40% des patient·es traité·es par luspatercept (exclusivement porteur·euses d’une mutation SF3B1 ou de sidéroblastes en anneau, réfractaires à l’EPO ou non éligibles) ont obtenu une indépendance transfusionnelle ≥8 semaines; la durée médiane de cette indépendance était comprise entre 24 et 42 semaines. Les effets indésirables les plus fréquents incluent céphalées, arthralgies, douleurs osseuses, fatigue, toux, douleurs abdominales, diarrhée et vertiges (principalement de grade 1/2).15
COMMANDS
Dans l’étude randomisée de phaseIII COMMANDS, 363 patient·es naïf·ves d’ESA (agents stimulant l’érythropoïèse), atteint·es d’anémie transfusion-dépendante et présentant un taux sérique d’érythropoïétine <500 U/l, ont été traité·es pendant au moins 24 semaines par luspatercept vs époétine alfa. Après un suivi médian d’environ 17 mois, 60% des patient·es traité·es par luspatercept ont atteint le critère d’évaluation primaire combiné d’indépendance transfusionnelle et d’augmentation de l’hémoglobine, contre 35% sous époétine alfa. De plus, 64% du groupe Luspatercept (42% sous EPO) ont atteint une absence de transfusion d’au moins 24 semaines.16
Dans les analyses posthoc de l’étude COMMANDS, l’avantage du luspatercept a été constant dans tous les sous-groupes étudiés (IPSS-R, taux d’EPO, besoins transfusionnels, statut mutationnel des sidéroblastes en anneau/SF3B1) et maintenu sur une période d’observation allant jusqu’à 24 mois. La durée médiane d’indépendance transfusionnelle était supérieure à 36 semaines dans tous les sous-groupes et le profil de sécurité n’a pas montré de nouveaux signaux à long terme.17 Le luspatercept permet à un plus grand nombre de patient·es de bénéficier d’une indépendance transfusionnelle à long terme (1,5 an sans transfusion: 30,2% vs 13,8%), la durée cumulée de tous les épisodes de réponse étant significativement plus longue (154,7 semaines vs 91,1 semaines).18
Le luspatercept est injecté par voie sous-cutanée toutes les trois semaines, généralement à une dose initiale de 1mg/kg, qui peut être progressivement augmentée jusqu’à 1,75mg/kg si la réponse de l’hémoglobine est insuffisante. La durée de réponse est nettement plus longue lorsque l’Hb ≥10g/dl est atteinte (199 vs. 27 semaines), ce qui plaide en faveur du rôle de l’escalade de dose.19 Des études évaluent déjà des doses initiales plus élevées de luspatercept.20
Les répondeur·euses au luspatercept bénéficient également d’un avantage en termes de survie globale. Par rapport à l’époétine alfa, outre une durée de réponse statistiquement et significativement plus longue, une tendance positive en faveur de la survie globale est observée de manière cohérente dans tous les sous-groupes (OS à 4,5 ans avec le luspatercept: 58,9% vs 41,8%).21
Imételstat
L’imételstat est un inhibiteur de la télomérase qui se lie de manière compétitive au composant ARN (hTR) de la télomérase et inhibe ainsi l’allongement des télomères dans les cellules progénitrices malignes. L’imételstat est le premier médicament de sa catégorie à avoir obtenu une autorisation de la FDA en 2024 et une autorisation conditionnelle de l’EMA en 2025 pour les patient·es adultes présentant une anémie transfusionnelle liée à un SMD non del(5q) à faible risque, qui ne répondent pas suffisamment à un traitement basé sur un ESA ou qui ne sont pas éligibles à ce traitement.
IMerge
Dans l’étude randomisée de phaseIII IMerge, 39,8% des patient·es traité·es par imételstat ont atteint une indépendance transfusionnelle ≥8 semaines contre 15% dans le groupe placebo, la durée médiane d’indépendance transfusionnelle étant supérieure à 30 semaines.22
L’imételstat est administré par perfusion intraveineuse toutes les quatre semaines; une prémédication ainsi que des contrôles réguliers de l’hémogramme et de la fonction hépatique sont nécessaires. Les effets indésirables très fréquemment observés sont des cytopénies de grade 3–4 (transitoires), une élévation des paramètres hépatiques, de la fatigue et des céphalées, généralement contrôlables par des adaptations posologiques.
Un avantage potentiel de l’imételstat par rapport au luspatercept pourrait concerner les patient·es fortement dépendant·es des transfusions. Des études supplémentaires sont attendues à ce sujet. Il convient de noter qu’en dépit de son autorisation en Europe, l’accès à la substance est actuellement limité.
Algorithmes possibles pour la décision thérapeutique
Lors du choix de l’agent thérapeutique, il convient tout d’abord de tenir compte de l’autorisation: à l’exception de l’EPO (pour l’anémie symptomatique avec Hb ≤10g/dl), un besoin transfusionnel est requis dans tous les cas pour l’instauration du traitement. Pour la suite de la sélection, le lénalidomide devrait être privilégié en raison des excellents résultats obtenus dans les SMD avec délétion isolée de del(5q). Pour toutes les autres entités, l’EPO (si une réponse est attendue selon le score nordique) ou, sur la base des données de l’étude COMMANDS, le luspatercept, s’imposent comme traitement de première ligne.
Les résultats de l’étude COMMANDS favorisent le luspatercept dans tous les sous-groupes; une différenciation éventuelle pourrait se faire en fonction du taux d’EPO et des besoins transfusionnels. Toutefois, de nombreux·ses patient·es qui ne sont pas encore dépendant·es des transfusion ont déjà reçu de l’EPO, de sorte que le luspatercept est souvent utilisé en deuxième ligne. L’imételstat constituerait ainsi une option prometteuse de deuxième ou troisième ligne.
Les recommandations actuelles d’Onkopedia23 (02/2024) n’intègrent pas encore les données récentes des études; une mise à jour est prévue pour 2026. La figure1 présente une procédure possible, modifiée selon une proposition italienne.24
%20%C3%A0%20faible%20risque.jpg)
Fig.1: Proposition d’algorithme thérapeutique pour l’anémie dans les SMD non del(5q) à faible risque (modifiée selon Santini V & Consagra A)24
Perspectives
Les succès obtenus jusqu’à ce jour stimulent la poursuite des recherches: de nouvelles substances reposant sur des mécanismes d’action totalement différents sont actuellement en développement clinique, et des thérapies combinées sont évaluées afin d’explorer d’éventuelles synergies. Il est également étudié si un traitement précoce de l’anémie associée aux SMD, avant même l’apparition d’une dépendance transfusionnelle, peut permettre d’obtenir de meilleurs résultats à long terme.
Conclusion
Il est possible d’éviter les transfusions sanguines dans les SMD à faible risque grâce à des thérapies ciblées, des stratégies modernes et une collaboration étroite entre médecins et patient·es. Une amélioration de la qualité de vie constitue un bénéfice avéré; une prolongation de la survie globale semble également possible. Les patient·es qui ne sont pas éligibles aux thérapies établies ou dont la maladie progresse malgré le traitement devraient, dans la mesure du possible, être inclus·es dans des études cliniques afin d’avoir accès à des substances innovantes.
Première publication en allemand dans JATROS Hämatologie & Onkologie 1/2026
Littérature:
1 Cazzola M: Myelodysplastic syndromes. N Engl J Med 2020; 383(14): 1358-74 2 Greenberg PL et al.: Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012; 120(12): 2454-65 3 Bernard E et al.: Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evidence 2022; 1(7): EVIDoa2200008 4 Pfeilstöcker M et al.: Time-dependent changes in mortality and transformation risk in MDS. Blood 2016; 128(7): 902-10 5 Fenaux P, Adès L: How we treat lower-risk myelodysplastic syndromes. Blood 2013; 121(21): 4280-6 6 Oliva EN et al.: Eltrombopag vs. placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): phase1 results of a single-blind, randomised, controlled, phase2 superiority trial. Lancet Haematol 2017; 4: e127-36 7 Passweg JR et al.: Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phaseIII trial comparing antithymocyte globulin plus cyclosporine with best supportive care – SAKK 33/99. J Clin Oncol 2011; 29: 303-9 8 Rathje K, Kröger N: Optimal timing of allogeneic hematopoietic stem cell transplant in MDS. Leuk Lymphoma 2025; 66(10): 1788-800 9 Bryant BJ et al.: Ascertainment of iron deficiency and depletion in blood donors through screening questions for pica and restless legs syndrome. Transfusion 2013; 53(11): 2683-91 10 Gattermann N: Iron overload in myelodysplastic syndromes (MDS). Int J Hematol 2018; 107(1): 55-63 11 Malcovati et al.: Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based prognostic scoring system (WPSS). Haematologica 2011; 96: 1433-40 12 Hellström-Lindberg E et al.: A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol 2003; 120: 1037-46 13 List AF et al.: Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med 2005; 352(6): 549-57 14 Park S et al.: Outcome of lower-risk patients with myelodysplastic syndromes without 5q deletion after failure of erythropoiesis-stimulating agents. Clin Oncol 2017; 35(14): 1591-7 15 Fenaux P et al.: Luspatercept in patients with lower-risk myelodysplastic syndromes. NEngl J Med 2020; 382(2): 140-51 16 Della Porta MG et al.: Luspatercept vs. epoetin alfa in erythropoiesis-stimulating agent-naive, transfusion-dependent, lower-risk myelodysplastic syndromes (COMMANDS): primary analysis of a phase 3, open-label, randomised, controlled trial. Lancet Haematol 2024; 11(9): e646-58 17 Garcia-Manero G et al.: Efficacy and safety of luspatercept vs. epoetin alfa in erythropoiesis-stimulating agent (ESA)-naive patients with transfusion-dependent lower-risk myelodysplastic syndromes (LR-MDS): full analysis of the COMMANDS trial. ASH 2023; Abstr. #193 18 Garcia-Manero G et al.: Long-term response analysis of transfusion independence in erythropoiesis stimulating agent–naive patients with very low-, low-, or intermediate-risk myelodysplastic syndromes treated with luspatercept vs epoetin alfa in the COMMANDS trial. ASH 2024; Abstr. #350 19 Santini V et al.: Clinical benefits of achieving hemoglobin (Hb) levels ≥10 g/dL in transfusion-dependent (TD) erythropoiesis-stimulating agent (ESA)-naive patients (pts) with lower-risk (LR) myelodysplastic syndromes (MDS) treated with luspatercept in the COMMANDS trial. ASH 2024; Abstr. #1818 20 DellaPorta G et al.: EHA 2025, Abstr. #PF634 21 Santini V et al.: Overall survival and duration of transfusion independence for first-line esa-naive patients with lower-risk myelodysplastic syndromes treated with luspatercept vs epoetin alfa in the COMMANDS trial. EHA 2025; Abstr. #S177 22 Platzbecker U et al.: Imetelstat in patients with lower risk myelodysplastic syndrome who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): a multinational, randomised, double blind, placebo controlled, phase 3 trial. Lancet 2024; 403: 249-60 23 Hofmann WK et al.: Myelodysplastische Neoplasien (Myelodysplastische Syndrome, MDS). Onkopedia-Leitlinie 2024. Online unter www.onkopedia.com 24 Santini V, Consagra A: How to use luspatercept and erythropoiesis-stimulating agents in low-risk myelodysplastic syndrome. Br J Haematol 2025; 207(1): 15-26
Das könnte Sie auch interessieren:

Gérer les effets indésirables de manière ciblée: pratique selon les directives S3 et internationales
Les thérapies systémiques contre le cancer prolongent la survie et permettent de contrôler les maladies tumorales. Il est déterminant que les patient·es tolèrent bien le traitement. Cela ...
Nouveaux résultats d’études à San Antonio
La prise en charge individualisée du carcinome mammaire est déjà très avancée dans la pratique clinique quotidienne. Néanmoins, des récidives tumorales surviennent et des patient·es ...
La radiothérapie adjuvante à l’ère des thérapies systémiques modernes
La radiothérapie adjuvante demeure un élément central dans le traitement des tumeurs malignes cutanées, malgré les progrès réalisés avec les immunothérapies et les thérapies ciblées. ...