
Weichteilsarkome – «Standard of Care»
Bei Weichteilsarkomen sind die genaue pathologische Diagnosestellung sowie die Diskussion der adäquaten Diagnostik und Therapie im Rahmen eines interdisziplinären Sarkomboardes an einem Sarkomzentrum immens wichtig. Während die vollständige chirurgische Resektion im lokalisierten Stadium massgebend für die Prognose ist, muss die Indikation einer additiven Radiotherapie und/oder Systemtherapie im Einzelfall geprüft werden.
Keypoints
-
Weichteilsarkome stellen eine heterogene Tumorgruppe von über 100 Subtypen dar, welche sich in Epidemiologie, klinischem Verlauf und Behandlung unterscheiden können.
-
Bei klinischem oder radiologischem Verdacht auf ein Sarkom sollte idealerweise noch vor der Biopsie eine Überweisung an ein Sarkomzentrum erfolgen. Die interdisziplinäre Falldiskussion an einem Sarkomboard beeinflusst die Prognose nachweislich.
-
Die vollständige Resektion des Tumors ist der entscheidendste Faktor für den Therapieerfolg. Eine additive Radiotherapie verbessert die lokale Kontrolle. Die Indikation einer neoadjuvanten/adjuvanten Chemotherapie wird kontrovers diskutiert.
-
Die palliative Systemtherapie verlängert das Gesamtüberleben. Eine Anthrazyklin-haltige Chemotherapie stellt in der ersten Linie den Standard dar.
Epidemiologie
Weichteilsarkome sind eine heterogene Tumorgruppe, bestehend aus mehr als 100 Subtypen.1 Aufgrund ihrer Inzidenz von 4,45/1000000 gehören sie definitionsgemäss zu den seltenen Tumorentitäten.2 Sie machen lediglich 1% der malignen Tumoren bei Erwachsenen aus, bei Kindern jedoch etwa 12%.3 Die Geschlechterverteilung ist in etwa gleich mit etwas höherer Inzidenz bei Männern. Das mittlere Erkrankungsalter liegt bei 62 Jahren. Die 5-Jahres-Überlebensrate hat sich in den letzten Jahren verbessert und beträgt 61,6%.2
Meistens treten Sarkome sporadisch auf. Zu den bekannten Risikofaktoren gehören gewisse syndromale Erkrankungen (z.B. Li-Fraumeni-Syndrom, Neurofibromatose), Exposition gegenüber Chemo- bzw. Radiotherapie, ein chronisches Lymphödem (Angiosarkome) sowie eine virale Infektion mit dem Humanen Herpesvirus Typ 8 (Kaposi-Sarkom).
Weichteilsarkome können ubiquitär im Körper auftreten. In ca. 30–50% entstehen sie jedoch an den Extremitäten. Neben vielen seltenen Subtypen mit wenigen Fällen jährlich lassen sich etwa 2/3 der Weichteilsarkome den drei häufigsten Subtypen, das bedeutet, den Liposarkomen, den Leiomyosarkomen und den undifferenzierten, pleomorphen Sarkomen zuordnen.2
Klinik, Diagnostik und Stadieneinteilung
Die Beschwerden sind abhängig von der Primärtumorlokalisation. Sarkome der Extremitäten fallen in der Regel über eine Schwellung mit oder ohne assoziierte Schmerzen auf. Die Symptome ergeben sich aus der Verdrängung bzw. Infiltration des gesunden Gewebes. Bei Diagnosestellung sind etwa 90% der Weichteilsarkome lokalisiert. Weichteilsarkome metastasieren selten lymphogen, meist hämatogen, wobei das Auftreten von Lungenmetastasen die weitaus häufigste Präsentation darstellt.4
Bei klinischem und/oder radiologischem Verdacht auf ein Weichteilsarkom sollte eine Überweisung an ein Sarkomzentrum ( www.sarkom-schweiz.ch ) erfolgen. Die Diskussion der Befunde und des diagnostischen und therapeutischen Vorgehens im Rahmen eines interdisziplinären Sarkom-Boards ist prognoserelevant.5,6
Die radiologische Diagnostik muss vor der Indikationsstellung einer etwaigen Biopsie erfolgen. Zur optimalen Darstellung des Primärtumors stellt die MRT den Standard zur Diagnostik eines Weichteilsarkoms der Extremitäten dar. Eine ergänzende konventionelle Aufnahme kann zusätzliche diagnostische Informationen bieten (z.B. Verkalkungen bei Synovialsarkomen). Bei retroperitonealen/abdominellen oder thorakalen Sarkomen genügt meist eine Computertomografie.
In Absprache mit dem Operateur wird eine Stanzbiopsie der Raumforderung geplant und durchgeführt.7 Die Richtung der Biopsie wird idealerweise so gewählt, dass der Biopsietrakt im Rahmen der Tumorresektion mitreseziert werden kann, um Implantationsmetastasen zu vermeiden. Von einer Feinnadelbiopsie wird aufgrund der geringen Grösse des Präparates und der damit verbundenen Schwierigkeit der Diagnosestellung abgeraten.
Das Staging wird in den meisten Fällen mittels Computertomografie durchgeführt, wobei in speziellen Fällen eine PET/CT hilfreich sein kann.4
Die prognostischen Stadien basieren auf der üblichen TNM-Klassifikation. Die meisten Weichteilsarkome werden histologisch gemäss Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) graduiert, welche den Grad der Tumordifferenzierung, die Mitoserate und den Anteil an Nekrosen im Primärtumor inkorporiert (Tab. 1).8 Zur Prognosebestimmung wurde kürzlich die «Sarculator»-App entwickelt. Dieses gibt uns basierend auf Tumor- und Patientencharakteristika validierte prognostische Informationen.9
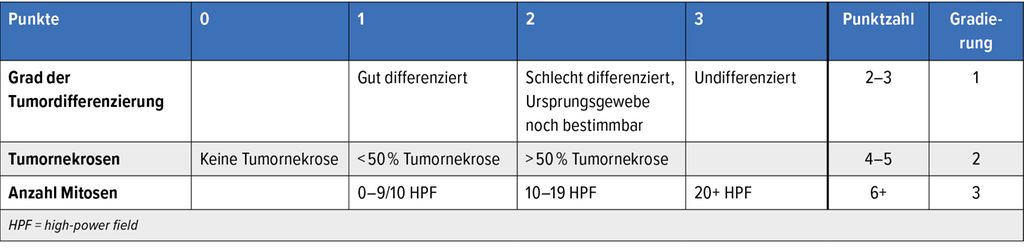
Tab. 1: Histologische Gradierung von Weichteilsarkomen gemäss FNCLCC (modifiziert nach Trojani M et al.)24
Therapie
Lokalisiertes Tumorstadium
Tumorresektion
Die Therapie der Wahl bei Weichteilsarkomen stellt die komplette chirurgische Exzision (R0) dar.10
Radiotherapie
Bei Weichteilsarkomen der Extremitäten mit hohem Rezidivrisiko (Grad 2–3, tiefe Lokalisation, >5cm) verbessert eine zusätzliche Radiotherapie die lokale Kontrolle, während der Nutzen in Bezug auf das Gesamtüberleben unklar ist. Hinsichtlich der Wirksamkeit besteht kein Unterschied, ob die Radiotherapie adjuvant oder neoadjuvant appliziert wird.11 Die Durchführung im neoadjuvanten Setting wird aufgrund des genau definierten Volumens bei in situ liegendem Tumor, der geringeren Dosis im Vergleich zur postoperativen Applikation und der geringeren posttherapeutischen funktionellen Einschränkungen durch etwaige Narbenbildungen favorisiert. Im Gegensatz zu den Extremitätensarkomen stellt eine additive Radiotherapie bei primär retroperitonealen Sarkomen keinen Standard dar.12
Systemtherapie
Der Nutzen einer adjuvanten Chemotherapie wird weltweit kontrovers diskutiert. Im Rahmen der letzten publizierten Metaanalyse wird eine absolute Risikoreduktion bzgl. des Gesamtüberlebens von 11% berichtet.13 Die grösste prospektiv-randomisierte Studie der EORTC (European Organisation for Research and Treatment of Cancer), welche in der Metaanalyse nicht eingeschlossen wurde, zeigte jedoch keinen Nutzen einer adjuvanten Chemotherapie.14 Retrospektive Analysen suggerieren einen Benefit bei höhergradigen (G3) und grösseren (>5cm) Tumoren sowie bei gewissen histologischen Subtypen. Die ESMO- und NCCN-Richtlinien empfehlen, die Vor- und Nachteile dieser Therapie mit dem Patienten zu diskutieren und den Entscheid gemeinsam zu fällen. Die Chemotherapie entspricht einer Kombinationschemotherapie mit einem Anthrazyklin und Ifosfamid. Die akuten Nebenwirkungen (Alopezie, ausgeprägte Hämatotoxizität, Kardiotoxizität) und die potenziellen Langzeitfolgen (Infertilität, Kardiotoxizität, Sekundämalignome) sind hierbei zu berücksichtigen.
Obwohl keine klinische Studie je den Nutzen einer neoadjuvanten Chemotherapie mit Placebo verglichen hat, hat der Stellenwert dieses Therapieansatzes in den letzten Jahren zugenommen. Gronchi et al.15 konnte in einer prospektiv-randomisierten Phase-III-Studie zeigen, dass eine Therapie mit Anthrazyklinen und Ifosfamid (A+I) für 3 Zyklen bei Hochrisiko- Sarkomen einer an den Tumorsubtyp angepassten Chemotherapie («histology-tailored»; HT) bei verschiedenen Sarkom-Subtypen der Extremitäten und des Körperstammes überlegen ist. In der finalen Analyse mit einem medianen Nachbeobachtungszeitraum von 52 Monaten war die geschätzte 5-Jahres-Gesamtüberlebensrate im A+I-Arm bei 76% gegenüber 66% im HT-Arm (HR: 1,77; 95% CI: 1,10–2,83; p=0,02). Die im Vergleich zu historischen Daten günstigeren Resultate, und dies im Vergleich mit einem aktiv anzunehmendem Therapiearm, dienen als Argumente für diese Indikation. Die prädiktive Bedeutung des Sarculator-Tools für den Einsatz einer (neo-)adjuvanten Systemtherapie wird basierend auf kürzlich publizierten retrospektiven Studien diskutiert.16
Die Verbesserung der Resektabilität insbesondere bei chemosensiblen Tumoren (z.B. undifferenzierte pleomorphe Sarkome, Synovialsarkom) stellt eine klarere Indikation dar.
Metastasiertes Tumorstadium
Die Zunahme der systemtherapeutischen Optionen hat in den letzten Jahren zu einer Verlängerung des Gesamtüberlebens von 12 auf ungefähr 20 Monate geführt. Die Wahl der Therapie ist von verschiedenen Patienten- und Tumorfaktoren (Alter, Komorbiditäten, Subtyp, Symptome etc.) abhängig. Der aktuelle Standard in der ersten Therapielinie ist Doxorubicin. Die Kombinationstherapie in Form von Doxorubicin und Ifosfamid ist gegenüber einer Monotherapie mit Doxorubicin in der Ansprechrate (ORR: 26% vs. 14%; p=0,0006) und im progressionsfreien Überleben (7,4 vs. 4,6 Monate; HR: 0,74)überlegen, jedoch nicht im Gesamtüberleben (14,3 vs. 12,8 Monate; HR: 0,83).17 Nach Versagen der Erstlinientherapie stehen folgende zugelassene Substanzen bereit: Trabectedin, Pazopanib, Eribulin und Dacarbazin. Es gilt wann immer möglich Patienten in Studien einzuschliessen.
Trabectedin ist zur Behandlung von Patienten mit Lipo- und Leiomyosarkom (L-Sarkome) nach Versagen oder Intoleranz von Anthrazyklinen und Ifosfamid zugelassen. Trabectedin konnte gegenüber «best supportive care» (BSC) in einer Phase-III-Studie primär bei L-Sarkomen eine signifikante Verlängerung des medianen PFS (5,1 vs. 1,4 Monate; HR: 0,29; p<0,0001), jedoch nicht des Gesamtüberlebens zeigen.18 Pazopanib, ein Multikinase-Inhibitor, wird ebenfalls zur Therapie bei Progression nach Anthrazyklin-haltiger Chemotherapie vergütet. Im Gegensatz zu Trabectedin erfolgte die Zulassung für alle Weichteilsarkome, mit Ausnahme von Liposarkomen. Pazopanib zeigte im Vergleich zu Placebo eine signifikante Verlängerung des medianen PFS, des primären Studienendpunktes (4,6 vs. 1,6 Monate, HR: 0,31; p<0,0001).19 Phase-II-Daten suggerieren eine Nichtunterlegenheit gegenüber Anthrazyklinen als Erstlinientherapie bei Patienten, die nicht für eine Chemotherapie qualifizieren.20,21 Eribulin kann in der häufigen Subgruppe der Liposarkome nach 2 Vortherapien als aktive Substanz eingesetzt werden.22 Im Rahmen einer prospektiven, randomisierten Phase-III-Studie wurde die Wirksamkeit von Eribulin bei L-Sarkomen mit Dacarbazin verglichen. Hierbei zeigte sich insbesondere in der Subgruppe der Liposarkome ein signifikanter Vorteil für das Gesamtüberleben unter Eribulin (15,6 vs. 8,4 Monate; HR: 0,51; 95% CI: 0,35–0,75; p<0,001).22 Immuntherapeutische Ansätze haben bisher in einer nicht selektionierten Sarkompopulation keine Erfolg versprechenden Resultate gezeigt. Der Stellenwert von Immuntherapien, u.a. in Kombination mit Tyrosinkinasehemmer in spezifischen Sarkomsubtypen, ist Gegenstand klinischer Forschung.
Nachsorge
Die Nachsorgeempfehlungen halten sich vornehmlich an internationale/nationale Konsensentscheidungen (Tab. 2). Leider fehlen zurzeit prospektive Studien, welche den prognostischen Nutzen von Nachsorgeuntersuchungen nach kurativ erfolgter Therapie unterstützen. Da im Falle eines primär pulmonalen, oligometastatischen Rezidivs ein geringes kuratives Therapiepotenzial besteht und im Verlauf ein Augenmerk auf therapieassoziierte Toxizitäten (z.B. Kardiotoxizität) gelegt werden muss, ist die Nachsorge dennoch sehr wichtig.
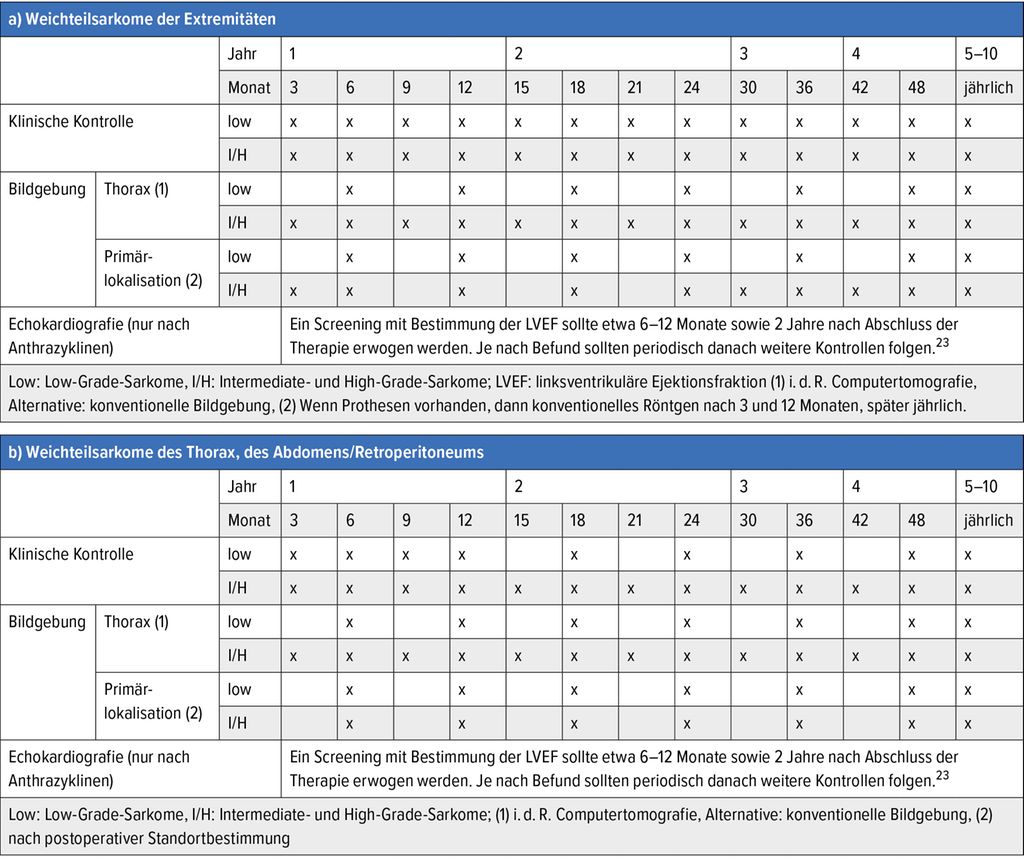
Tab. 2: Nachsorgeschema chirurgisch behandelter Patienten (modifiziert nach Swiss National Sarcoma Advisory Board)25
Literatur:
1 WHO classification of soft tissue and bone tumours; 2020 (5th edition) 2 Kollar A et al.: Incidence, mortality, and survival trends of soft tissue and bone sarcoma in Switzerland between 1996 and 2015. Cancer Epidemiol 2019; 63: 101596 3 Siegel RL et al.: Cancer Statistics, 2021. CA Cancer J Clin 2021; 71(1): 7-33 4 Christie-Large M et al.: Imaging strategy for detecting lung metastases at presentation in patients with soft tissue sarcomas. Eur J Cancer 2008; 44(13): 1841-5 5 Ray-Coquard I et al.: Sarcoma: concordance between initial diagnosis and centralized expert review in a population-based study within three European regions. Ann Oncol 2012; 23(9): 2442-9 6 Blay JY et al.: Improved survival using specialized multidisciplinary board in sarcoma patients. Ann Oncol 2017; 28(11): 2852-2859 7 Strauss DC et al.: The role of core needle biopsy in the diagnosis of suspected soft tissue tumours. J Surg Oncol 2010; 102(5): 523-9 8 Amin, MB et al.: The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more «personalized» approach to cancer staging. CA Cancer J Clin 2017; 67(2): 93-9 9 The «Sarculator»: http://www.sarculator.com 10 Gronchi A et al.: Extremity soft tissue sarcoma in a series of patients treated at a single institution: local control directly impacts survival. Ann Surg 2010; 251(3): 506-11 11 Casali PG et al.: Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018; 29(Suppl 4): iv51-iv67 12 Bonvalot S et al.: Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 2020; 21(10): 1366-1377 13 Pervaiz N et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008; 113(3): 573-81 14 Woll PJ et al.: Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): amulticentre randomised controlled trial. Lancet Oncol 2012; 13(10): 1045-54 15 Gronchi A et al.: Neoadjuvant chemotherapy in high-risk soft tissue sarcomas: final results of a randomized trial from italian (ISG), spanish (GEIS), french (FSG), and polish (PSG) sarcoma groups. J Clin Oncol 2020; 38(19): 2178-2186 16 Pasquali S et al.: The impact of chemotherapy on survival of patients with extremity and trunk wall soft tissue sarcoma: revisiting the results of the EORTC-STBSG 62931 randomised trial. Eur J Cancer 2019; 109: 51-60 17 Judson I et al.: Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: arandomised controlled phase 3 trial. Lancet Oncol 2014; 15(4): 415-23 18 Le Cesne A et al.: A randomized phase III trial comparing trabectedin to best supportive care in patients with pre-treated soft tissue sarcoma: T-SAR, aFrench Sarcoma Group trial. Ann Oncol 2021; 32(8): 1034-44 19 van der Graaf WT et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012; 379(9829): 1879-86 20 Grünwald V et al.: Randomized Comparison of pazopanib and doxorubicin as first-line treatment in patients with metastatic soft tissue sarcoma age 60 years or older: results of a german intergroup study. J Clin Oncol 2020; 38(30): 3555-64 21 Hirbe AC et al.: A phase II study of pazopanib as front-line therapy in patients with non-resectable or metastatic soft-tissue sarcomas who are not candidates for chemotherapy. Eur J Cancer 2020; 137: 1-9 22 Schöffski P et al.: Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016.; 387(10028): 1629-37 23 Curigliano G et al.: Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol 2020; 31(2): 171-90 24 Trojani M et al.: Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 1984; 33(1): 37-42 25 Swiss National Sarcoma Advisory Board: Recommendations for follow-up and surveillance. Online unter: https://www.swiss-sarcoma.net/pdf/GCP_12_sarcoma_follow-up_guideline.pdf
Das könnte Sie auch interessieren:
Adjuvantes Osimertinib reduziert ZNS-Rezidive bei EGFR-mutierter Erkrankung
Etwa 30% der Patienten mit nicht kleinzelligem Lungenkarzinom (NSCLC) präsentieren sich mit resezierbarer Erkrankung und werden einer kurativen Operation unterzogen. Viele Patienten ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...