Myeloproliferative Neoplasien: ein Update vom EHA-Meeting 2020
Das EHA-Meeting 2020 stand dieses Jahr ganz im Zeichen der aktuellen Coronakrise. Wie viele andere wissenschaftliche Großveranstaltungen wurde das konventionelle EHA-Meeting, das heuer in Frankfurt hätte stattfinden sollen, durch ein virtuelles Meeting ersetzt. Erfreulicherweise hatte dieser Umstand keinen Einfluss auf die hohe wissenschaftliche Qualität der Beiträge. Frühe klinische Studien mit innovativen Therapieansätzen scheinen im Bereich der Philadelphia-negativen myeloischen Neoplasien (MPN) eine neue Phase im Management dieser Krankheitsgruppe einzuleiten. Berichte über molekulare Aberrationen des RAS-Signalpfades dominieren Studien zur Pathophysiologie fortgeschrittener Ph-negativer MPN.
Keypoints
-
Mit KRT-232, Parsaclisib und Tagraxofusp stehen bei fortgeschrittenen myeloischen Neoplasien drei neue Substanzen zur Verfügung, die in frühen klinischen Studien vielversprechende Ergebnisse zeigen.
-
Eine Überaktivierung des RAS-Signalpfads bei myeloproliferativen Neoplasien führt zu einer Veränderung des hämatologischen Phänotyps der Erkrankung und zu einer Verschlechterung ihrer Prognose.
-
Eine therapeutische Beeinflussung des Inflammasoms könnte geeignet sein, die klinischen Konsequenzen einer RAS-Pfad-vermittelten Myeloproliferation zu verbessern.
Klinische Studien bei chronischer myeloischer Leukämie
Asciminib ist ein neuer, spezifischer Inhibitor von BCR-ABL, der das Onkoprotein im Bereich seiner Myristoyl-bindenden Tasche beeinflusst. Dieser einzigartige Wirkmechanismus begründet die Hoffnung auf ein verbessertes Sicherheitsprofil gegenüber den derzeit verfügbaren ATP-bindenden Tyrosinkinase-Inhibitoren (TKI). In einer von T. Hughes präsentierten Studie wurden Patienten mit chronischer myeloischer Leukämie (CML) mit Asciminib mit dem Ziel eines verbesserten Ansprechens behandelt, die nach mindestens zwei TKI refraktär, im Rezidiv oder intolerant waren und die zwar auf einen TKI eine gewisse, aber keine tiefe Response gezeigt hatten.1 Der Ausgangswert von BCR-ABL musste bei ≤1% liegen, Patienten mit einer T315I-Mutation wurden ausgeschlossen. Sechs Patienten (12,5%) beendeten bisher die Therapie wegen unerwünschter Effekte, 42 Patienten (87,5%) sind nach wie vor unter Therapie, 36 (75%) haben mindestens eine „major molecular response“ (MMR) erreicht. Die häufigsten (>10%) Nebenwirkungen des Grades 3/4 des Prüfpräparats waren Anstiege der Lipase (27,1%) und Hypertonie (12,5%). Bei Patienten ohne MMR, MR4 oder MR4,5 zum Ausgangszeitpunkt kam es zu einem kontinuierlichen Anstieg des kumulativen Ansprechens, sogar über 48 Wochen hinaus mit Ansprechraten von 75% MMR, 42,1% MR4 und 42,9% MR4,5. Die mediane Zeit bis zum Ansprechen betrug 30 Tage. Alle 18 Patienten, die eine MMR erreichten, behielten dieses Ansprechen für mehr als zwei Jahre.
Das Absetzen eines TKI bei CML-Patienten mit sehr gutem molekularem Ansprechen wurde in der Vergangenheit bereits in kontrollierten klinischen Studien untersucht, aber Daten zum Absetzen eines TKI bei CML-Patienten in der Real-World-Situation sind kaum verfügbar. In Schweden wurden >95% der Patienten mit CML in einem nationalen Register erfasst. Zwischen 2007 und 2012 wurde bei 584 Patienten die Diagnose CML gestellt, Informationen betreffend Unterbrechung einer TKI-Therapie sind von 548 Patienten verfügbar, die in einer Studie von H. Flygt präsentiert wurden.2 Bei 234 Patienten wurde eine Unterbrechung der TKI-Behandlung für mehr als einen Monat berichtet. Die Gründe für diese Unterbrechung waren ein tiefes molekulares Ansprechen bei 56%, Nebenwirkungen bei 18%, eine allogene Stammzelltransplantation bei 13% und andere Gründe bei 12%. 29% der Patienten beendeten den TKI im Rahmen einer klinischen Studie, 70% der Patienten beendeten ihre TKI-Therapie außerhalb einer Studie. Zum Zeitpunkt der Auswertung erfolgte eine Reinitiierung der TKI-Therapie bei 48,9% der Patienten nach einer medianen Zeit von 0,4 Jahren. Diese Untersuchung belegt, dass TKI im klinischen Alltag bereits abgesetzt werden. Die Wahrscheinlichkeit für solche Patienten, auch weiterhin behandlungsfrei zu bleiben, beträgt nach 22 Monaten 61%.
Klinische Studien bei Ph-negativen myeloproliferativen Neoplasien
MDM2 ist ein Negativregulator des Tumorsuppressorproteins p53, der in zirkulierenden Stamm-/Progenitorzellen überexprimiert ist. Patienten mit Myelofibrose (MF) sind durch eine massive Expansion des Progenitorzell-Kompartiments im Blut gekennzeichnet. In einer von H.K. Al-Ali präsentierten Studie wurde mit KRT-232 ein potenter, oraler Inhibitor von MDM2 bei Patienten mit MF untersucht.3 Es handelte sich dabei um eine einarmige, offene, multizentrische Phase-II-Studie bei Patienten mit MF, die sich nach vorangegangener JAK-Inhibitor-Therapie im Rezidiv befanden oder refraktär waren. 82 Patienten wurden in einen der drei Studienarme eingebracht, KRT-232 120mg (A1) oder 240mg (A2) täglich an den Tagen 1–7 eines 21-Tage-Zyklus oder 240mg (A3) täglich an den Tagen 1–7 eines 28-Tage-Zyklus. Endpunkt dieser Studie war eine mittels MRT oder CT gemessene Verringerung des Milzvolumens um >35% in Woche 24. Abbildung 1 zeigt einen Wasserfallplot der unterschiedlichen Therapiearme.
KRT-232 zeigte ein dosisabhängiges Verhältnis von Sicherheit und Wirksamkeit und eine vielversprechende Aktivität bei MF-Patienten in einer Hochrisikosituation. Durch diese „Proof of concept“-Studie wurde eine Dosis von 240mg KRT-232 täglich an den Tagen 1–7 eines 28-Tage-Zyklus für den zweiten Teil dieser Studie festgelegt.
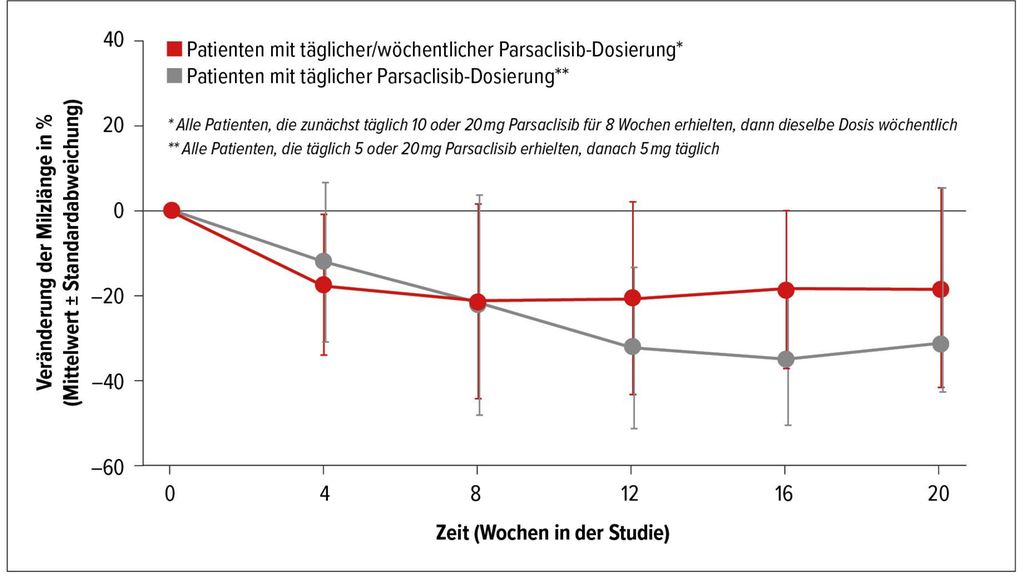
Eine weitere Pathophysiologie-basierte Strategie bei Patienten mit MF und suboptimalem Ansprechen auf Ruxolitinib wäre die medikamentöse Hemmung des PI3K/AKT-Signalpfades, die sich schon bei anderen Tumorentitäten als wirksamer Behandlungsansatz erwiesen hat. Parsaclisib ist ein hochselektiver PI3Kδ-Inhibitor der nächsten Generation. Patienten mit primärer oder sekundärer MF und suboptimaler Response auf Ruxolitinib erhielten in einer von A. Yacoub präsentierten Studie neben Ruxolitinib Parsaclisib in einer Dosis von 10 bzw. 20mg täglich für acht Wochen und anschließend einmal wöchentlich, oder 5mg bzw. 20mg Parsaclisib täglich für acht Wochen und 5mg täglich danach.4 Der primäre Endpunkt der Studie war eine Änderung des Milzvolumens gegenüber dem Ausgangsbefund in Woche 12. In Abbildung 2 sind die mittleren Veränderungen des Milzvolumens (dargestellt als Milzlänge) in beiden Behandlungsgruppen dargestellt.
Der Symptomenscore nach zwölf Wochen zeigte eine Verminderung um minus 14% bzw. minus 51%. Die Zugabe von Parsaclisib zeigte also eine eindeutige Wirksamkeit bei Patienten mit MF und suboptimalem Ansprechen auf Ruxolitinib alleine.
Tagraxofusp ist ein Interleukin(IL)-3-Molekül, das mit einem trunkierten Diphtherietoxin fusioniert ist. Diese Substanz kommt daher als neue therapeutische Strategie für IL-3-Rezeptor-exprimierende myeloische Neoplasien infrage und wurde bereits für die Behandlung des blastischen plasmazytoiden dendritischen Neoplasmas zugelassen. In einer multizentrischen, offenen Phase-I/II-Studie, die von N. Pemmaraju vorgestellt wurde, wurde dieses Medikament als 15-minütige Infusion in einer Dosis von 7, 9 oder 12μg/kg an den Tagen 1–3 im Rahmen von vier dreiwöchigen Behandlungszyklen und anschließend im Rahmen vier weiterer 28-tägiger Behandlungszyklen verabreicht.5 32 Patienten mit Hochrisiko-MF wurden im Rahmen dieser Studie behandelt, inklusive 13 Patienten, die mindestens drei Vortherapien erhalten hatten. Von 19 auswertbaren Patienten zeigten 53% eine Reduktion der Milzgröße, vier davon erreichten eine Verkleinerung der Milz um >45%. Interessanterweise zeigten vier von fünf Patienten mit Splenomegalie und Monozytose ein Milzansprechen, zwei davon um >45%. Die Responserate bei Symptomen betrug 46%. Die am häufigsten beobachteten Nebenwirkungen (>15%) inkludierten Kopfschmerzen, Hypalbuminämie, erhöhte Transaminasen, Thrombozytopenie und Anämie. Die häufigsten Nebenwirkungen ≥ Grad 3 waren Thrombopenie (8%) und Anämie (15%). Die Studie zeigt, dass Tagraxofusp als Monotherapie einen klinischen Benefit erzeugt und ein vorhersehbares und handhabbares Sicherheitsprofil aufweist.
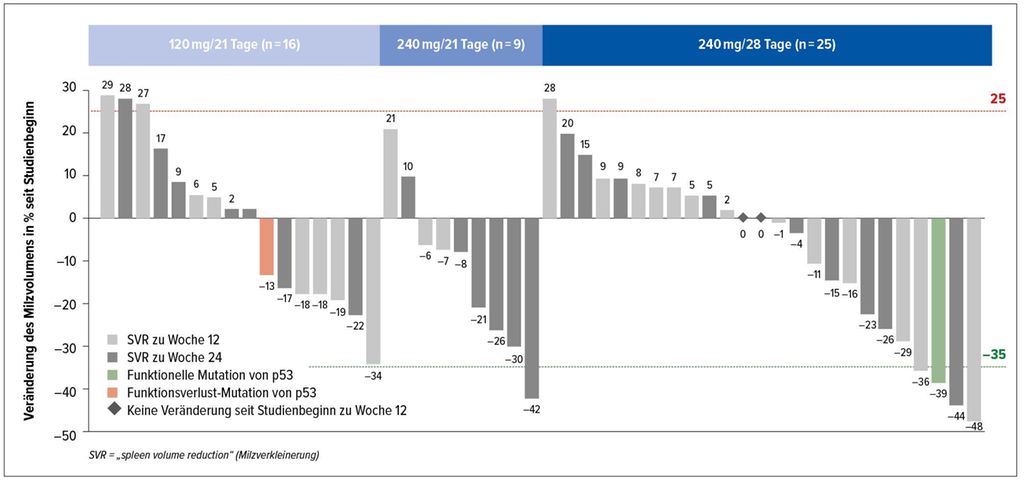
Abb. 1: Veränderung des Milzvolumens unter KRT-232 (nach Al-Ali HK et al.)3
Myeloproliferative Neoplasien: wichtige pathophysiologische Erkenntnisse
Eine hochinteressante Arbeit von S. Hamarsheh gibt neue Einblicke in die Rolle des RAS-Pfades in der Pathophysiologie von myeloproliferativen Neoplasien.6 Unter Verwendung eines Mausmodells mit induzierbarer KRAS-Mutation wurde die funktionelle Rolle des Inflammasoms für die RAS-Pfad-assoziierte Myeloproliferation untersucht. Dazu wurden Mäuse mit KRAS-Aktivierung und NLRP3-Defizienz generiert. Während Mäuse mit KRASMyeloproliferation und Zytopenie entwickelten, wurden diese Veränderungen bei Mäusen, denen NLRP3 fehlte, nicht beobachtet. Auch die therapeutische Gabe des IL-1-Rezeptorantagonisten Anakinra verminderte die Myeloproliferation bei KRAS-aktiven Mäusen. Die Ergebnisse im Mausmodell konnten auch in Zellproben von Patienten mit chronischer myelomonozytärer Leukämie (CMML), juveniler myelomonozytärer Leukämie (JMML) und akuter myeloischer Leukämie (AML) reproduziert werden. Diese Daten eröffnen neue therapeutische Möglichkeiten durch NLRP3-Blockade bei KRAS-mutierten myeloischen Neoplasien.
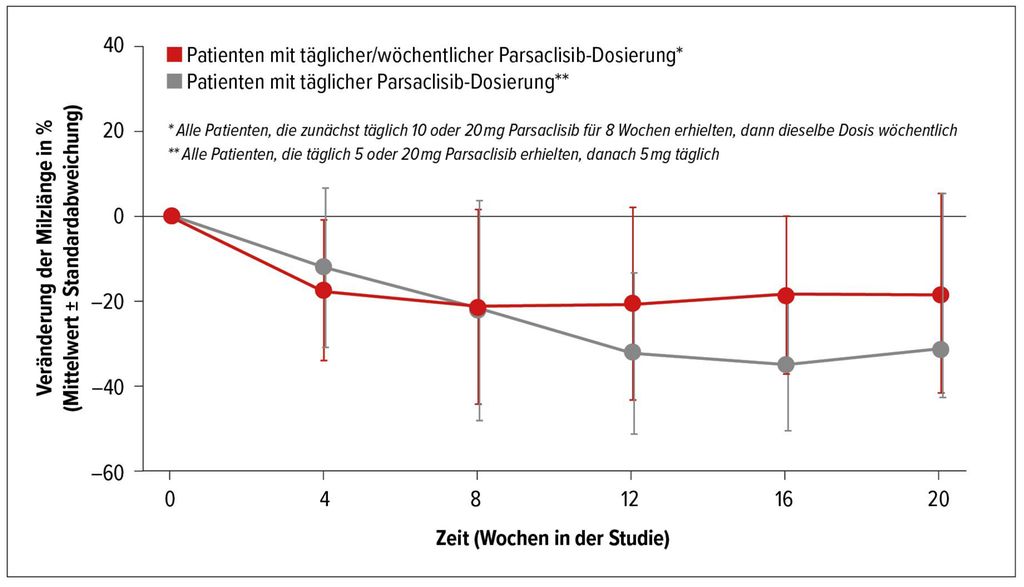
Abb. 2: Veränderung der Milzlänge unter Parsaclisib (nach Yacoub A et al.)4
In einer weiteren Studie wurde von M.Meggendorfer eine „Whole exome“-Analyse bei 19 MPN-Patienten vorgestellt, bei denen Zellmaterial sowohl von der chronischen Phase als auch vom Zeitpunkt der Progression vorlag.7 Neben den Treibermutationen in JAK2 (89%) und MPL (11%) waren Mutationen in SRSF2 (68%), TET2 (47%), ASXL1 (26%), RUNX1, DNMT3A, ZRSR2 (je 16%) sowie SETBP1 und IDH2 (je 11%) nachweisbar. Im Durchschnitt gingen während der Progression der Erkrankung neun Mutationen verloren, während zwölf neu entstanden. Am häufigsten zeigte sich ein Zugewinn von RUNX1 (n=6) und TP53 (n=4). Interessanterweise verloren 37% (7/19) Patienten ihre MPN-definierende Mutation (JAK2 oder MPL), während Mutationen in der Splicing-Maschinerie und in den Chromatin-modifizierenden Genen während der Krankheitsprogression stabil blieben. Eine funktionelle Annotation ergab, dass der Anteil von Patienten mit einer Mutation in RAS-Pfad-bezogenen Genen im Zuge der Krankheitsprogression von 58% (11/19) auf 74% (14/19) anstieg.
Mutationen im RAS/MAPK-Pfad wurden auch bei 464 konsekutiven Patienten mit WHO-definierter MF mittels NGS von G. Coltro untersucht.8 RAS/MAPK-Pfad-Gene umfasstenNRAS, KRAS und CBL. Insgesamt wurden 53 Patienten (11,4%) identifiziert, bei denen mindestens eine Mutation im RAS-Signalpfad gefunden wurde. Die mediane VAF-Frequenz von 36% war deutlich geringer als jene der Treibermutationen. RAS-Pfad-Mutationen waren mit 20% wesentlich häufiger bei primärer MF als bei Patienten mit präfibrotischer MF (8%) und sekundärer MF (6%). Im Vergleich zu Wildtyp-Patienten waren Patienten mit RAS-Pfad-Mutationen älter, hatten höhere Leukozytenwerte, ein niedrigeres Hämoglobin, eine geringere Thrombozytenzahl, höhere Blasten im Blut, mehr Transfusionsabhängigkeit und konstitutionelle Symptome. In einer univariaten Analyse hatten RAS-Pfad-mutierte Patienten ein kürzeres Survival, das seine Signifikanz auch in einer multivariaten Analyse unter Einbeziehung von MIPSS-Scores und Zytogenetik behielt. Eine Transformation in eine AML war mit 28% in der RAS-mutierten Kohorte ebenfalls häufiger als in der Wildtyp-Kohorte (10%; p<0,0001). Bei 61 Patienten, die eine JAK-Inhibitor-Therapie erhielten, konnte ein Milzansprechen beurteilt werden. Der Anteil von Patienten, die eine Milzresponse zeigten, war in der RAS-Pfad-mutierten Kohorte signifikant geringer als in der Wildtyp-Kohorte (9% vs. 70%; p=0,0002). Zusammenfassend sind Mutationen im RAS-Pfad nicht selten und mit einem veränderten Phänotyp sowie kurzem Überleben assoziiert. Außerdem dürfte der Nachweis einer RAS-Mutation auf ein vermindertes Ansprechen auf JAK2-Inhibitoren hinweisen.
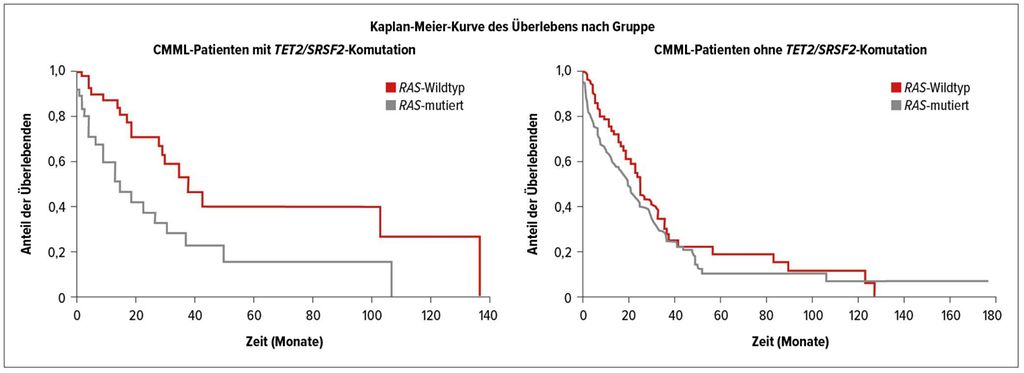
Abb. 3: Überleben bei TET2/SRSF2-Komutation (nach Geissler K et al.)9
Bei unselektierten Patienten mit CMML wird der prognostische Einfluss von RAS-Pfad-Mutationen noch unterschiedlich betrachtet, weil nur in einem Teil der Studien ein signifikanter negativer Effekt auf das Überleben dieser Patienten nachgewiesen werden konnte. Eine Erklärung für die unterschiedlichen Ergebnisse in den Analysen könnten die beträchtlichen Unterschiede in den Mutationsprofilen dieser Patienten sein. Eine Komutation von TET2 und SRSF2 ist eine häufige und weitgehend spezifische Veränderung bei CMML-Patienten. Wie eine von K. Geissler präsentierte Auswertung der Austrian Biodatabase for CMML (ABCMML) ergab, zeigten 87 von 291 molekular untersuchten Patienten eine solche Komutation.9 Bei 37 Patienten fanden sich zusätzliche Mutationen in einer der RAS-Signalpfad-Komponenten. Patienten mit zusätzlichen RAS-Pfad-Mutationen hatten höhere Leukozytenwerte, niedrigere Hämoglobinspiegel, geringere Thrombozytenwerte sowie häufiger Splenomegalie und Zeichen der Transformation. Das Überleben dieser Patienten war mit 15 Monaten signifikant kürzer als jenes der Patienten mit einer TET2/SRSF2-Komutation ohne RAS-Pfad-Veränderungen (Abb.3).
Interessanterweise gab es bei CMML-Patienten ohne TET2/SRSF2-Komutation keinen signifikanten Unterschied im Überleben der RAS-positiven und RAS-negativen Patienten. Mit dieser Untersuchung konnte gezeigt werden, dass in einer molekular definierten Subgruppe von CMML-Patienten eine Überaktivierung des RAS-Signalpfads mit Veränderungen des klinisch-hämatologischen Phänotyps und mit einem deutlich kürzeren Überleben assoziiert ist. Diese Ergebnisse unterstreichen die große pathophysiologische Bedeutung des RAS-Signalpfads bei der myeloproliferativen Form der CMML.
Literatur:
1 Hughes T et al.: Asciminib in heavily pretreated patients (pts) with Philadelphia-chromosome-positive (PH+) chronic myeloid leukemia in chronic phase (CML-CP) sensitive to tyrosine kinase inhibitor (TKI) therapy. EHA 2020, Abstr. #S170 2 Flygt H et al.: High level of successful TKI discontinuation outside clinical trials - a population-based study from the Swedisch CML registry. EHA 2020, Abstr. #S174 3 Al-Ali HK et al.: KRT-232, a first-in-class, murine double minute 2 inhibitor (MDM2I), for myelofibrosis (MF) relapsed or refractory (R/R) to janus-associated kinase inhibitor (JAKI) treatment (TX). EHA 2020, Abstr. #S215 4 Yacoub A et al.: Addition of parsaclisib, a PI3Kdelta inhibitor, in patients (pts) with suboptimal response to ruxolitinib (RUX): aphase 2 study in pts with myelofibrosis (MF). EHA 2020, Abstr. #S216 5 Pemmaraju N et al.: Updated results from a phase 1/2 clinical trial of tagraxofusp, a CD123-targeted therapy, in patients with poor-risk myelofibrosis. EHA 2020, Abstr. #S218 6 Hamarsheh S et al.: Oncogenic KRAS-G12D causes myeloproliferation via NLRP3 inflammasome activation. EHA 2020, Abstr. #S212 7 Meggendorfer M et al.: Exome sequencing of paired MPN and blast phase shows an accumulation of splicing and chromatin modifying gene mutations, clonal evolution and gain of RAS pathway mutations during progression. EHA 2020, Abstr. #S213 8 Coltro G et al.: RAS/MAPK pathway mutations are associated with adverse survival outcomes and may predict resistance to JAK inhibitors in myelofibrosis. EHA 2020, Abstr. #S211 9 Geissler K et al.: Impact of RAS-pathway activation on phenotype and outcome in patients with chronic myelomonocytic leukemia and a TET2/SRSF2 comutation. EHA 2020, Abstr. #EP783
Das könnte Sie auch interessieren:
Adjuvantes Osimertinib reduziert ZNS-Rezidive bei EGFR-mutierter Erkrankung
Etwa 30% der Patienten mit nicht kleinzelligem Lungenkarzinom (NSCLC) präsentieren sich mit resezierbarer Erkrankung und werden einer kurativen Operation unterzogen. Viele Patienten ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...