Die Genetik von gastrointestinalen Stromatumoren
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Gastrointestinale Stromatumoren (GIST) stellen die häufigsten mesenchymalen Tumoren im Magen-Darm-Trakt dar. Mit einer Inzidenz in Deutschland von ca. 6 Betroffenen pro 100 000 Einwohner ist jährlich mit etwa 5000 Neuerkrankungen zu rechnen. Das Verständnis ihrer onkogenen Pathogenese hat GIST zu einem Paradigma für die Relevanz von Tyrosinkinasen und deren Inhibition in soliden Tumoren gemacht.
Keypoints
-
Der Mutationsstatus bei GIST ist prognoserelevant und prädiktiv für das Therapieansprechen auf Tyrosinkinaseinhibitoren.
-
Histologischer Phänotyp und Genotyp korrelieren – spindelzellige Tumoren tragen oft KIT-Mutationen, GIST mit epitheloider Morphologie PDGFRA-Mutationen.
-
Die Zahl der alternativ alterierten Gene in GIST ohne KIT- oder PDGFRA-Mutation ist bei vermehrtem Einsatz der Tiefensequenzierung erheblich gestiegen. So sind Mutationen in BRAF, KRAS, HRAS, NRAS, PIK3CA, ARID1A/B, FGFR1, MEN1 und anderen Genen beschrieben sowie kasuistisch erwähnte Translokationen wie z.B. ETV-NTRK3, FGFR1-HOOK3, FGFR1-TACC1, KIT-PDGFRA und PRKAR1B-BRAF.
-
Bei mehr als einem GIST in einer Familie (syn- oder metachron) muss eine hereditäre Disposition ausgeschlossen werden.
Seit der Erstbeschreibung von aktivierenden Mutationen in der Rezeptortyrosinkinase KIT durch Hirota et al. 19981 konnten weitere relevante Gene identifiziert werden, die vielfach ein anderes biologisches Verhalten und Therapieansprechen der betroffenen Tumoren zur Folge haben. Zumeist handelt es sich um sporadische, nur im Tumorgewebe nachweisbare Mutationen, in 10 bis 15% der Fälle liegen hingegen hereditäre bzw. syndromale Alterationen vor.
Da die genomische Lokalisation und Art der Mutation prognosebestimmend und zudem therapierelevant sind, ist die Mutationsanalyse in der Mehrzahl der GIST bereits bei Erstdiagnose anzustreben. Es folgt ein Überblick über die Häufigkeit der unterschiedlichen genomischen Alterationen und deren klinische Bedeutung.
Sporadische GIST sind am häufigsten in KIT oder PDGFRA mutiert
Bei Einsatz hochsensitiver Sequenziermethoden kann in etwa 73% der Fälle mit einer KIT-Mutation gerechnet werden, davon gut 62% in KIT Exon 11 und knapp 9% in KIT Exon 9 (lt. GIST-Register Münster mit 3837 Fällen). Die weiteren betroffenen Genabschnitte in KIT (Exone 8, 13, 14 und 17) machen jeweils etwa 1% oder weniger aus.
Das mit ca. 14% am zweithäufigsten mutierte, im Jahr 2002 erstmalig in GIST als mutiert beschriebene Gen ist PDGFRA,2 welches große Sequenzhomologien zu KIT aufweist und direkt neben diesem auf Chromosom 4 liegt. Das PDGFRA-Protein besitzt funktionell analoge Domänen wie das KIT-Protein, in diesen zeigt sich aber eine deutlich andere Verteilung der Alterationen. KIT- und PDGFRA-Primärmutationen schließen sich gegenseitig aus.
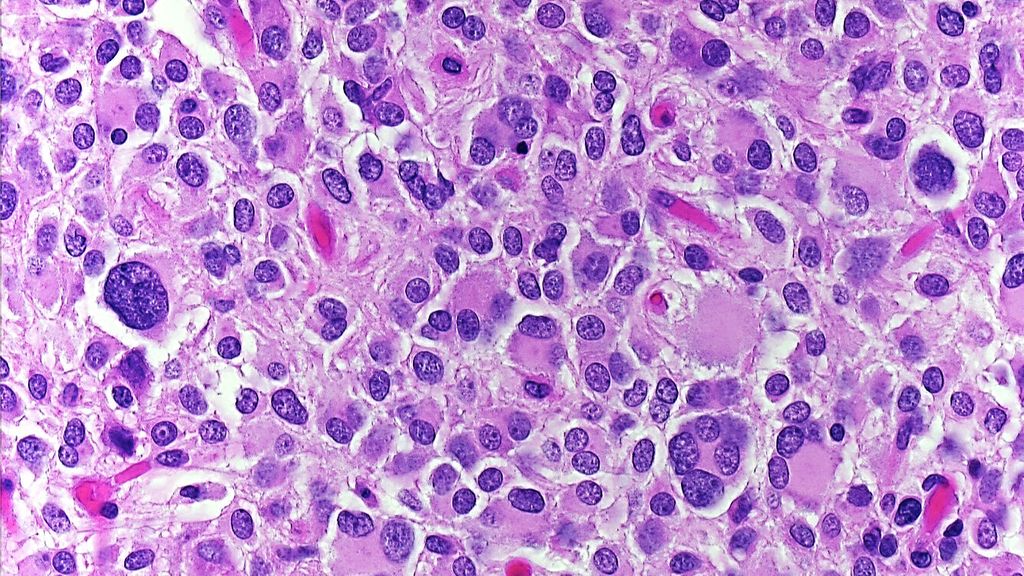
Hinsichtlich ihrer Lokalisation und Prognose verhalten sich PDGFRA-mutierte GIST anders als KIT-mutierte. Erstere sind fast ausschließlich im Magen lokalisiert, während Letztere auch im übrigen Gastrointestinaltrakt vorkommen. Die beiden genomischen GIST-Hauptgruppen unterscheiden sich aber nicht nur biologisch sondern auch morphologisch. PDGFRA-mutierte GIST sind vielfach epitheloidzellig3,4 im Gegensatz zu den überwiegend spindelzelligen KIT-mutierten Tumoren (Abb. 1a und b).
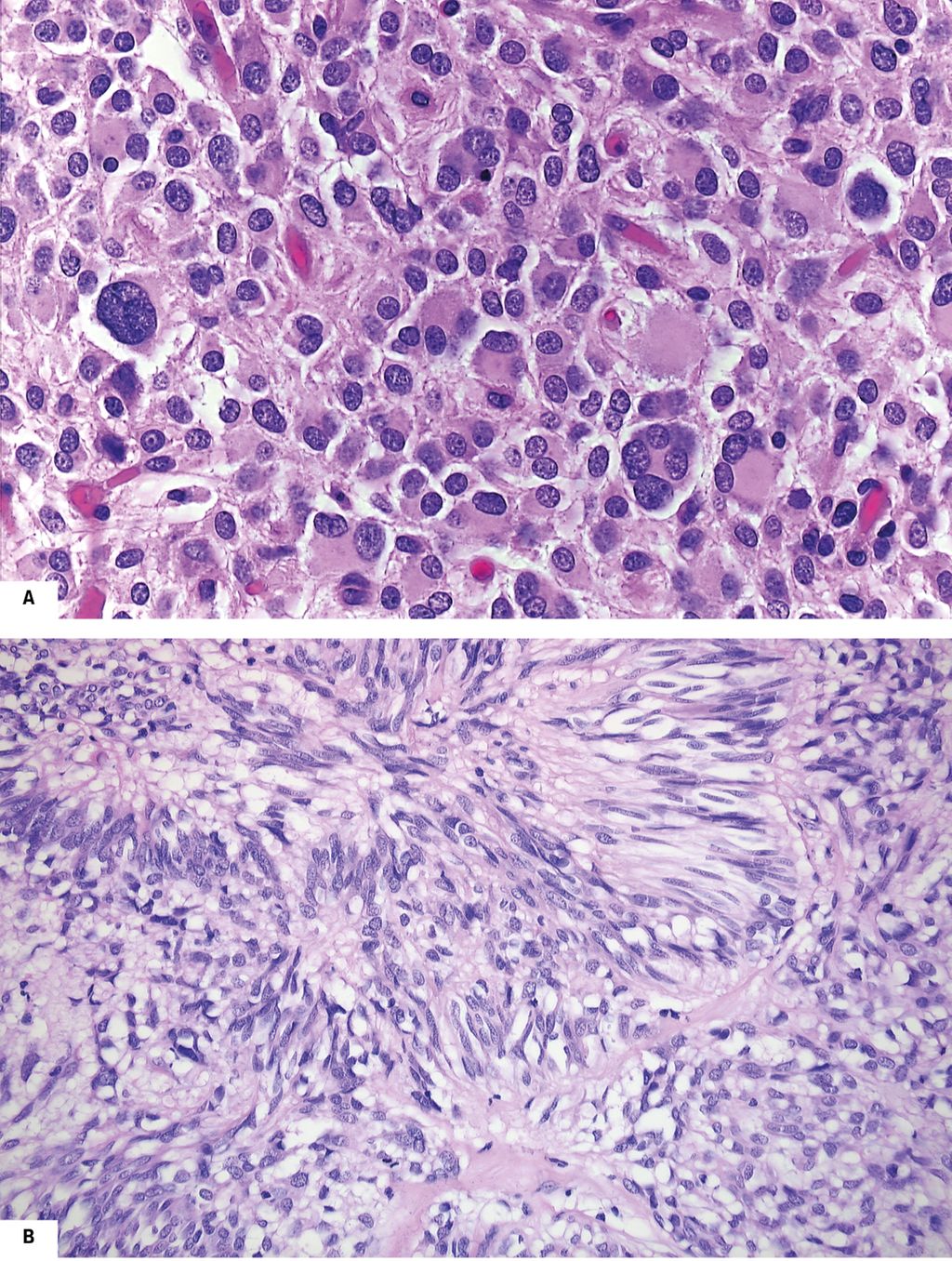
Abb. 1: Unterschiedliche histologische Subtypen bei KIT- bzw. PDGFRA-mutierten GIST (jeweils HE, Originalvergrößerung x200 in A, x100 in B). A) Epitheloidzelliger GIST mit PDGFRA-Mutation. B) Spindelzelliger GIST mit KIT-Mutation
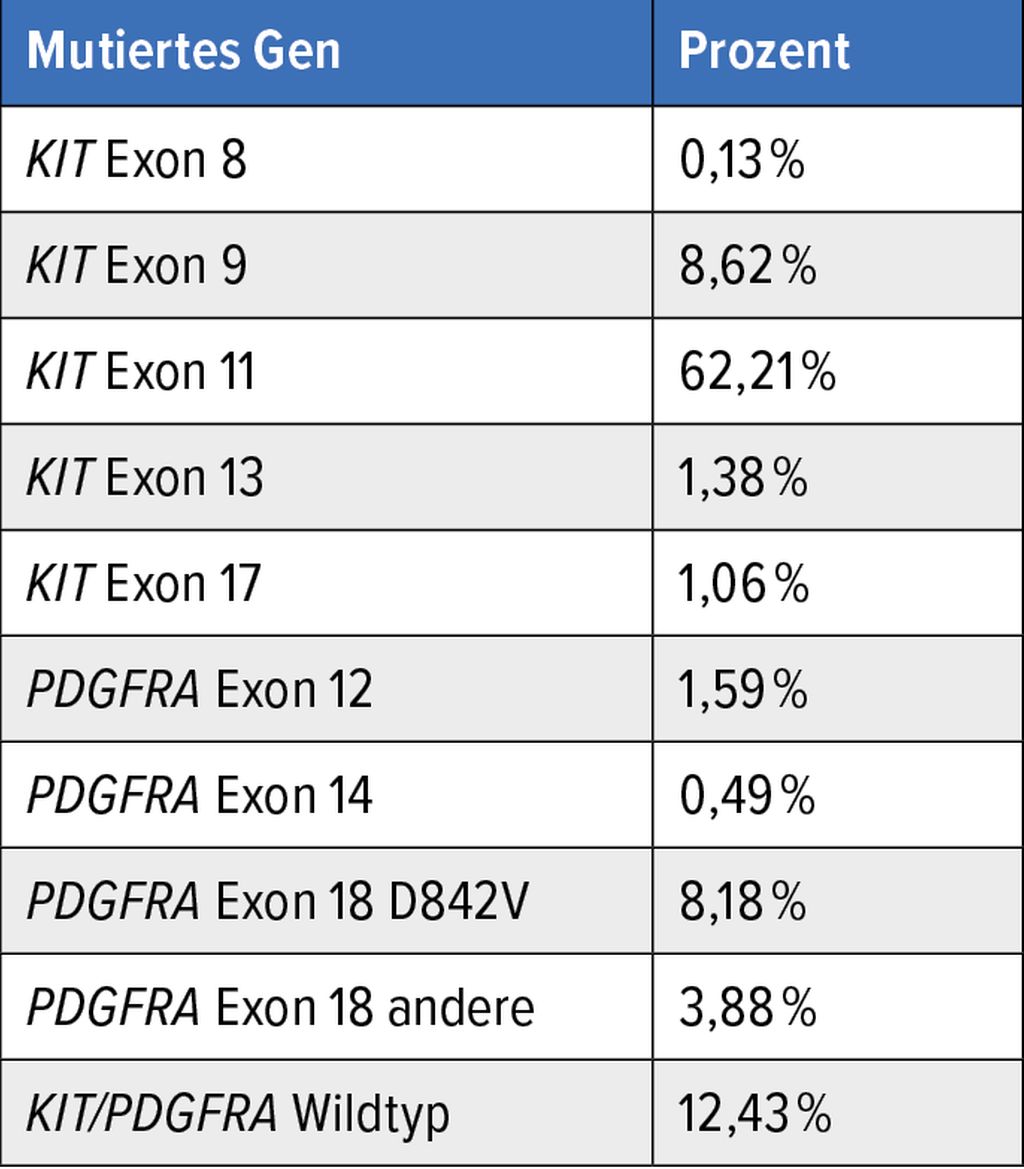
Tab. 1: Lokalisation und Häufigkeit von KIT/PDGFRA-Mutationen, n=3837 (Daten des GIST-Registers Münster)
Prognostisch sind PDGFRA-mutierte GIST mehrheitlich als günstiger einzustufen, was vermutlich auf ihre geringere Proliferationsaktivität zurückzuführen ist.5 Bei Vergleich der Mutationstypen in beiden Genen dominieren in KIT membrannahe Genabschnitte (Exon 11 und 9), in PDGFRA hingegen Mutationen in Tyrosinkinasedomänen (Exon 18 und selten Exon 14). Die Mutationshäufigkeiten in beiden Genen im Münsteraner GIST-Register sind in Tabelle 1 aufgeführt.
KIT-/PDGFRA-mutierte GIST zeigen ein unterschiedliches Ansprechen auf Tyrosinkinaseinhibitoren
Neben der unterschiedlichen Biologie und Morphologie beider GIST-Hauptgruppen sind diese auch unterschiedlich sensitiv bezüglich einer Therapie mit dem Goldstandard der medikamentösen Behandlung, dem Tyrosinkinaseinhibitor Imatinib.6 Das beste Ansprechen auf Imatinib weisen KIT-Exon-11-mutierte Tumoren auf, während KIT-Exon-9-mutierte GIST ein intermediäres Ansprechen zeigen und daher mit einer verdoppelten Tagesdosis Imatinib (800mg/Tag) behandelt werden.7,8 Dies gilt nicht nur für metastasierte Tumoren, sondern findet mittlerweile auch in der adjuvanten Therapie Anwendung mit einer signifikanten Reduktion des Rückfallrisikos.9
Bei den PDGFRA-mutierten GIST tritt in zwei Drittel der Fälle die Punktmutation p.Asp842Val (p.D842V) auf, die eine primäre Imatinib-Resistenz zur Folge hat. Erst seit diesem Jahr ist mit Avapritinib erstmals ein bei diesem Mutationstyp wirksamer Tyrosinkinaseinhibitor für inoperable oder metastasierte GIST zugelassen.10
GIST ohne KIT-/PDGFRA-Mutation sind durch andere genomische oder epigenetische Alterationen gekennzeichnet
Bei etwa 12% aller GIST lässt sich weder in KIT noch in PDGFRA eine Mutation nachweisen. Als erster Schritt empfiehlt sich in dieser Situation zunächst die Wiederholung der Mutationsanalyse in einem zweiten Labor, da insbesondere lange KIT-Deletionen mittels „next-generation sequencing“ (NGS) übersehen werden können. Bei Wildtypsequenzen in KIT und PDGFRA sind vielfach Alterationen in anderen Genen nachzuweisen.
Die größte Gruppe stellen hier GIST mit Succinatdehydrogenase(SDH)-Ausfall dar. Dieser Komplex aus vier Isoformen ist Bestandteil des mitochondrial lokalisierten Citratzyklus. Bei Fehlfunktion einer der vier Isoformen verliert der SDH-Komplex seine Fähigkeit, Succinat in Fumarat zu überführen. In der Folge kommt es zu einer Akkumulation von Succinat im zellulären Zytoplasma, welches zu einer Aktivierung des Transkriptionsfaktors HIF1-alpha führt.
Ein derartiger Funktionsausfall kann entweder bei Keimbahnmutationen in einem der SDH-Gene (A, B, C und D) oder auch sporadisch auftreten.11
Schließlich sind sehr seltene Mutationen im Neurofibromatose-1-Gen (sporadisch und hereditär), in BRAF, KRAS, HRAS, NRAS, PIK3CA, ARID1A/B, FGFR1, MEN1 und in weiteren Genen beschrieben, daneben finden sich in der Literatur kasuistisch erwähnte Fusionstranskripte wie z.B. ETV-NTRK3, FGFR1-HOOK3, FGFR1-TACC1, KIT-PDGFRA und PRKAR1B-BRAF.12
Hereditäre GIST haben diverse Pathogenesen
Bislang sind etwa 40 Familien weltweit mit einer Keimbahnmutation in KIT oder PDGFRA beschrieben. Mehrheitlich handelt es sich um heterozygote Keimbahnmutationen im KIT-Gen, während bislang lediglich vier Familien und eine Einzelperson mit analoger PDGFRA-Mutation beschrieben wurden (siehe Ricci).13 Bei erstgenannter Konstellation kann auch von einem KIT-Mutationssyndrom, bei Zweiterem von einem PDGFRA-Mutationssyndrom gesprochen werden.
Eine weitere hereditäre Konstellation stellt die Neurofibromatose Typ 1 (NF1) dar, bei der es neben der Entwicklung von Neurofibromen und ZNS-Tumoren auch zu einem gehäuften Auftreten von GIST kommt. Das Risiko von NF1-Patienten, an einem oder mehreren GIST zu erkranken, steigt im Laufe des Lebens kontinuierlich an.14
Schließlich kommt es wie oben beschrieben bei Keimbahnmutationen in einem der SDH-Gene (A, B, C und D) zu einer hohen Inzidenz von GIST, häufig kombiniert mit Paragangliomen (Carney-Stratakis-Syndrom).15 Bei Defizienz von SDHC liegt zumeist eine epigenetische Regulationsstörung vor, die zu einer Koinzidenz von GIST, Paragangliomen und pulmonalen Chondromen führt und Carney-Triade genannt wird.13;16–17 Unabhängig davon, welche Isoform der SDH-Gene genetisch oder epigenetisch alteriert ist, kommt es in der Regel zu einem immunhistochemischen Ausfall von SDHB, was in der pathologischen Diagnostik von GIST sehr hilfreich ist.18
Eine hereditäre Genese ist dringend zu vermuten, wenn es in der Familie des Patienten mindestens eine weitere an GIST erkrankte Person gibt oder eine systemische Mastozytose oder Paragangliome in der Familie kursieren. Auch Zeichen der Neurofibromatose Typ 1 sollten als Warnsignal für ein mögliches gehäuftes Auftreten von GIST berücksichtigt werden.
Literatur:
1 Hirota S et al.: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279: 577-80 2 Heinrich MC et al.: PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708-10 3 Wardelmann E et al.: Association of platelet-derived growth factoralphamutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn 2004; 6(3): 197-204 4 Pauls K et al.: PDGFRA- and c-kit mutated gastrointestinal stromal tumors (GISTs) are characterized by distinctive histological and immunohistochemical features. Histopathology 2005; 46(2): 166-75 5 Miettinen M, Lasota J: Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23: 70-83 6 Demetri JD et al.: Efficacy and safety of Imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472-80 7 Verweij J et al.: Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomized trial. Lancet 2004; 364: 1127-34 8 Heinrich MC et al.: Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB150105 Study by Cancer and Leukemia Group Band Southwest Oncology Group. J Clin Oncol 2008; 26(33): 5360-67 9 Joensuu H et al.: Twelve vs. 36 months of adjuvant imatinib as treatment of operable GIST with a high risk of recurrence: final results of a randomized trial (SSGXVIII/AIO). JAMA 2012; 307: 1265-72 10 Heinrich MC et al.: Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (navigator): A multicentre, open-label, phase 1 trial. Lancet Oncol 2020; 21: 935-46 11 Haller F et al.: Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer 2014; 21(4): 567-77 12 Brčić I et al.: Update on molecular genetics of gastrointestinal stromal tumors review diagnostics (Basel). 2021; 11(2): 194 13 Ricci R: Syndromic gastrointestinal stromal tumors. Hereditary Cancer in Clinical Practice 2016; 14: 15 14 Miettinen M et al.: Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006; 30: 90-6 15 Pasini B et al.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet 2008; 16: 79-88 16 Carney J, Stratakis C: Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Gen 2002; 108: 132-9 17 Zhang L et al.: Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol 2010; 34: 53-64 18 Gaal J et al.: SDHB immunohistochemistry: a useful tool in the diagnosis of Carney – Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol 2011; 24: 147-51
Das könnte Sie auch interessieren:
Adjuvantes Osimertinib reduziert ZNS-Rezidive bei EGFR-mutierter Erkrankung
Etwa 30% der Patienten mit nicht kleinzelligem Lungenkarzinom (NSCLC) präsentieren sich mit resezierbarer Erkrankung und werden einer kurativen Operation unterzogen. Viele Patienten ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...