
Das ungelöste Problem vom richtigen Zeitpunkt
Bei AL-Amyloidosen im Rezidiv ist es derzeit völlig unklar, welcher Zeitpunkt am besten für den Start einer Rezidivtherapie geeignet ist.
Keypoints
-
Kontinuierliche Kontrolle auch in Zeiten der Remission ist unbedingt notwendig.
-
NTproBNP und kardiale Beteiligung zum Zeitpunkt der Diagnose sind die einzigen unabhängigen Parameter für das mediane Überleben im AL-Rezidiv. Das heißt, NTproBNP immer bei Kontrolle bestimmen.
-
Die Rezidivtherapie sollte gestartet werden, bevor es zu einem Anstieg des NTproBNP kommt.
-
High-Risk-dFLC-Progression gilt als Biomarker-Konstellation, die den Beginn einer Rezidivtherapie signalisiert.
Diagnosekriterien und Therapieindikation zum Zeitpunkt der Diagnose
Die Diagnose eines multiplen Myeloms (MM) wie auch einer AL-Amyloidose (AL) wird meistens nicht zufällig gestellt, sondern aufgrund von Symptomen, die durch Organschäden hervorgerufen werden. Beim multiplen Myleom werden die Organschäden hauptsächlich durch die deregulierte Plasmazellproliferation verursacht. Bei den AL-Amyloidosen werden die Organschäden durch das toxische Paraprotein ausgelöst. Die Diagnosen werden mithilfe klarer Diagnosekriterien erstellt.
Seit 2003 werden für die Sicherung der Diagnose von multiplen Myelomen die CRAB-Kriterien herangezogen. Es müssen dabei mindestens 10% klonale Plasmazellen in der Knochenmarkbiopsie sowie eine Biopsie-gesicherte Myelomläsion des Knochens oder einer extramedullären Manifestation nachweisbar sein. Zusätzlich muss zumindest ein Organschaden vorliegen, der klar durch die Plasmazellerkrankung hervorgerufen wird. Die häufigsten Organschäden werden als CRAB-Kriterien zusammengefasst.1 Um ein Fortschreiten des Schadens zu verhindern, besteht zum Zeitpunkt der Diagnose eine klare Therapieindikation. 2014 wurden zusätzlich die SLIM-Kriterien2 definiert, die einen früheren Therapiestart ermöglichen, bevor es zur Ausbildung von irreparablen Schäden kommt.
Um eine AL zu diagnostizieren, müssen ebenfalls klare Kriterien erfüllt sein.3 Es muss eine klonale Plasmazelldyskrasie mit einem nachweisbaren Paraprotein (Knochenmark, Serum und/oder Harn) vorliegen. Außerdem muss es eine Gewebebiopsie geben, an der das Vorliegen einer Leichtkettenamyloidose eindeutig nachgewiesen werden kann. Auch bei der Diagnose einer AL gilt, dass umgehend eine Therapie eingeleitet werden muss, um das Fortschreiten des Organschadens zu verhindern.
Rezidivkriterien und Therapie-indikation im Rezidiv
Die Situation im Rezidiv ist völlig anders. Aufgrund der vorbekannten Erkrankung werden regelmäßige Kontrollen durchgeführt, auch wenn die Patienten eine komplette Remission erreicht haben. Die allermeisten Rezidive werden daher in einem sehr frühen Stadium als rein biochemisches Rezidiv erfasst. Das toxische Paraprotein kann in minimalen Konzentrationen nachgewiesen werden, ohne dass dadurch bereits ein neuerlicher Organschaden ausgelöst wird. Das gilt sowohl für multiple Myelome wie auch für AL-Amyloidosen. Es stellt sich daher die Frage: Ab wann sollte ein Rezidiv behandelt werden? Sofort beim Wiederauftritt des Paraproteins in der Phase des biochemischen Rezidivs oder wenn erste Zeichen von Organschäden auftreten? Ein Argument für Zweiteres wäre, dass durch die regelmäßigen Kontrollen auch die Organschäden in einem sehr frühen Stadium erkennbar wären. Die Frage, wann ein Rezidiv behandelt werden soll, ist im Verlauf einer AL-Amyloidose von besonderer Bedeutung, da sich die wiederauftretenden Organschäden anscheinend über einen längeren Zeitraum entwickeln.
2005 wurde anlässlich des 10. Meetings der Internationalen Amyloidose Gesellschaft ein Consensus-Statement zu Diagnose-, Response- und Rezidiv-Kriterien verfasst.4 In dieser Publikation wurden erstmals die Definition einer AL-Amyloidose-Rezidiv-Erkrankung bzw. die Kriterien einer Progression festgelegt.
Progression ausgehend von einer kompletten hämatologischen Remission:
-
Jede Form eines neuerlich messbaren monoklonalen Paraproteins
-
Neuerlich positive Immunfixation unabhängig davon, ob im Serum oder Harn vorhanden
-
Anstieg des freien Leichtketten-Levels von normal zu abnormal
-
Verdoppelung der Konzentration der Amyloid-formenden freien Leichtketten
Progression ausgehend von einer partiellen Remission:
-
50% Anstieg des M-Proteins im Serum auf 0,5g/dl (5g/l)
-
50% Anstieg des M-Proteins im Harn auf 200mg/Tag
-
Ein sichtbarer M-Spike in der Elektrophorese
-
Anstieg der Levels an freien Leichtketten auf 50% auf >10mg/dl (100mg/l)
Ein Statement dazu, wann mit einer Rezidiv-Therapie gestartet werden sollte, ist in dem Consensus-Stament nicht enthalten.
2018 wurde dieses schwierige Thema in einer Publikation von Palladini et al. erneut aufgegriffen.5 Basierend auf der Einführung von neuen, sehr aussagekräftigen Biomarkern und immer besser wirksamen therapeutischen Optionen bei der AL wurde das Thema der Progressionsklassifizierung neuerlich in Angriff genommen. Auch die Evaluierung des besten Zeitpunkts für den Beginn einer Rezidivtherapie wurde analysiert. In dieser Studie wurden 259 konsekutive Patienten untersucht, die auf ihre initiale Therapie angesprochen und im Verlauf ihrer Erkrankung ein Rezidiv erlitten hatten. 32 Patienten verstarben. 92 (35%) Patienten benötigten eine Rezidivtherapie. Das mediane Überleben nach Beginn der Rezidivtherapie der gesamten Kohorte lag bei 59 Monaten. Das mediane Überleben nach Rezidivtherapie war unabhängig davon, ob die ursprüngliche Induktionstherapie wieder aufgegriffen wurde oder ein anderes Therapieregime angewandt wurde.
Bei 20 Patienten mit ursprünglich kardialer Beteiligung wurde die Rezidivtherapie erst nach Anstieg der Differenz der freien Leichtketten (dFLC) und des NTproBNP gestartet. Das mediane Überleben lag in dieser Patientengruppe bei nur 17 Monaten. Ein Anstieg von NTproBNP und eine kardiale Beteiligung bei Diagnose konnten als unabhängige Parameter für das Überleben von Patienten mit Rezidiv eruiert werden.
Zusätzlich konnte in dieser Analyse eine Parameterkonstellation herausgearbeitet werden, die als Hochrisiko-Pattern gelten könnte: High-Risk-dFLC-Progression bestehend aus:
-
einer Differenz zwischen involvierten und nicht involvierten freien Leichtketten (dFLC) von > 20 mg/l,
-
einem Level von freien Leichtketten von 20 % des Ausganswertes und
-
einem 50%igen Anstieg des Werts zum Zeitpunkt des besten Ansprechens.
Diese High-Risk-dFLC-Progression konnte bei 85% der Patienten etwa 6 Monate vor der kardialen Verschlechterung nachgewiesen werden. Insgesamt ergaben die Daten, dass die Rezidivtherapie unbedingt vor Eintreten einer kardialen Verschlechterung eingeleitet werden sollte. Die High-risk-dFLC-Progressionsparameter könnten dabei als gute Indikatoren für einen raschen Therapiebeginn verwendet werden. Diese Ergebnisse müssen allerdings noch an einer größeren Patienten-Kohorte validiert werden, es wäre aber ein wirklich großer Fortschritt in der AL-Rezidiv-Therapie. Bis dahin bleibt das Problem des richtigen Zeitpunktes für den Start einer Rezidivtherapie weiterhin ungelöst.
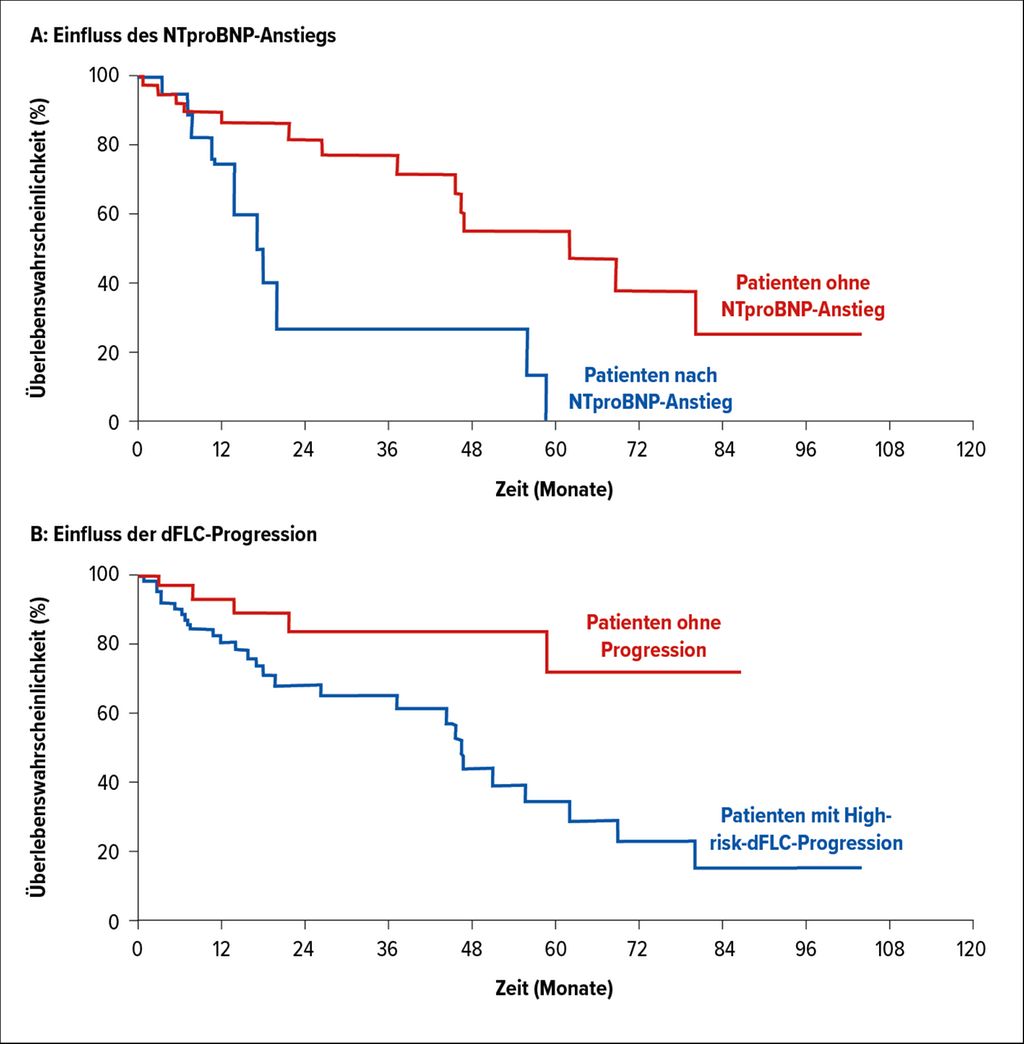
Abb. 1: Einfluss auf das Überleben (OS) im Rahmen der Rezidivtherapie: (A) Einfluss des NTproBNP-Anstiegs, (B) Einfluss der dFLC-Progression (mod. nach Palladini G et al.)5
Literatur:
1 International Myeloma Working Group: Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5): 749-57 2 Rajkumar SV et al.: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12): e538-48 3 Gavriatopoulou M et al.: European myeloma network recommendations on diagnosis and management of patients with rare plasma cell dyscrasias. Leukemia 2018; 32(9): 1883-98 4 Gertz MA et al.: Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 2005; 79(4): 319-28 5 Palladini G et al.: Presentation and outcome with second-line treatment in AL amyloidosis previously sensitive to nontransplant therapies. Blood 2018; 131(5): 525-32
Das könnte Sie auch interessieren:
Adjuvantes Osimertinib reduziert ZNS-Rezidive bei EGFR-mutierter Erkrankung
Etwa 30% der Patienten mit nicht kleinzelligem Lungenkarzinom (NSCLC) präsentieren sich mit resezierbarer Erkrankung und werden einer kurativen Operation unterzogen. Viele Patienten ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...