ILD: Das therapeutische Portfolio wächst
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Lange war die interstitielle Lungenerkrankung (ILD) bei systemischer Sklerose (SSc) weitgehend unbehandelbar. Das hat sich deutlich geändert, denn inzwischen stehen verschiedene Therapien zur Wahl. Darüber hinaus lassen sich nicht mehr nur krankheitsbedingt entstandene Schäden beobachten, sondern auch die Krankheitsaktivität quantifizieren. All dies unterstützt aktives Screening und frühe Intervention.
Keypoints
-
Verschiedene RCT liefern Evidenz für die Wirksamkeit einiger Therapeutika gegen SSc-ILD wie MMF, Cyclophosphamid, Nintedanib (FDA-Zulassung 4/2020), Tocilizumab (FDA-Zulassung 3/2021).
-
Es werden neue Marker benötigt, die vorzugsweise die Krankheitsaktivität statt des bereits bestehenden Schadens anzeigen, ein erster ist die FAPI-PET-CT.
-
Es bleibt zu beantworten, ob unterschiedliches Ansprechen von Haut und Lunge auf verschiedenen Wirkmechanismen beruht oder eine Frage des Messparameters ist, welcher Patient welche Therapie bekommen sollte, worauf Synergien beruhen und ob die sogenannte subklinische interstitielle Lungenerkrankung (in jedem Fall) behandelt werden muss.
Im Praxisalltag präsentierten sich interstitielle Lungenerkrankungen als diverses Spektrum, das von rein pneumologischen Diagnosen wie der idiopathischen pulmonalen Fibrose (IPF) über Pneumokoniosen bis zur Sarkoidose reiche, erklärte Prof. Dr. Jörg Distler, Düsseldorf, anlässlich seines Vortrag am letztjährigen DGRh-Kongress. Bei einem knappen Fünftel der Betroffenen sei die ILD mit einer rheumatologischen Grunderkrankung assoziiert. Den Großteil machten rheumatoide Arthritis und systemische Sklerose (SSc) aus, Letztere mit einem besonders hohen Anteil progredienter Verläufe. Daneben gebe es die Synthetase- und Mischkollagenose-assoziierten interstitiellen Lungenerkrankungen.1
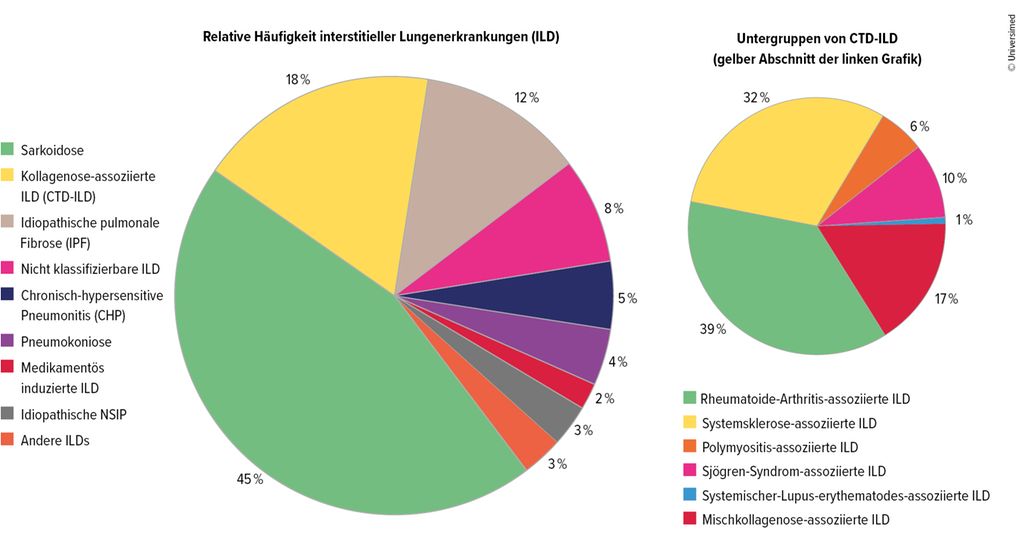
Abb. 1: Kollagenose-assoziierte ILD (CTD-ILD) machen 18 % der ILD aus und sind in weitere Untergruppen unterteilt. Mod. nach Wijsenbeek M et al.1
Man unterscheide eine diffuse kutane von einer limitierten kutanen Form der SSc. Bei der diffusen kutanen Form gingen die fibrotischen Veränderungen proximal über Ellenbogen oder Knie hinaus, während sie bei der limitierten kutanen distal auf Ellenbogen bzw. Knie begrenzt seien. Wie alle Studien bestätigten, gehe die diffuse kutane Erkrankung mit einem signifikant höheren Risiko für eine interstitielle Lungenerkrankung einher als die limitierte kutane Form. Je nach Studienkohorte und Screening-Tool erreiche die ILD-Inzidenz bei limitierter kutaner SSc 20 bis 50%, bei diffuser kutaner SSc 40 bis 75%. Röntgenuntersuchungen erbrächten geringere Werte als die HRCT.
Die ILD manifestiere sich meist früh im SSc-Verlauf, innerhalb der ersten 5 Jahre, danach stiegen die Manifestationen wesentlich langsamer. In neueren Datenanalysen verschöben sie sich zeitlich nach hinten, was der früher beginnenden und teilweise aggressiveren Behandlung mit neuen Medikamenten geschuldet sein könne.2–4
Prognose und Risikofaktoren
Die ILD sei auch im Hinblick auf die Prognose relevant, betonte Distler. Eine Analyse zur SSc-assoziierten Mortalität anhand von Daten von 1972 bis 2000 habe gezeigt, dass in den 70er-Jahren rund 5% der Patientinnen und Patienten mit SSc an interstitieller Lungenerkrankung starben, im Jahr 2000 war dies bereits die dominante Todesursache mit über 30%. Gleichzeitig seien die Todesfälle infolge einer „scleroderma renal crisis“ (SRC) signifikant zurückgegangen: von 40 auf 5%. Auch neuere Daten der inzwischen weltweit agierenden Forschungsgemeinschaft EUSTA zur diffusen bzw. limitierten kutanen SSc bestätigten die ILD als dominante Todesursache – und damit ihre prognostische Relevanz.5, 6
Bestünden bereits respiratorische Symptome wie eine Dyspnoe, sei dies mit einer höheren ILD-Inzidenz verknüpft, ebenso eine Raucheranamnese und gewisse Ethnien (z.B. afroamerikanisch). Außerdem hätten Männer und Menschen mit diffuser kutaner SSc häufiger schwerer verlaufende interstitielle Lungenerkrankungen, gefördert durch Anti-Topoisomerase-1-Antikörper, die mit der diffusen kutanen SSc assoziiert sind. Die primär mit der limitierten kutanen Form assoziierten Anti-Zentromer-Antikörper senkten hingegen das individuelle Risiko für eine ILD.7
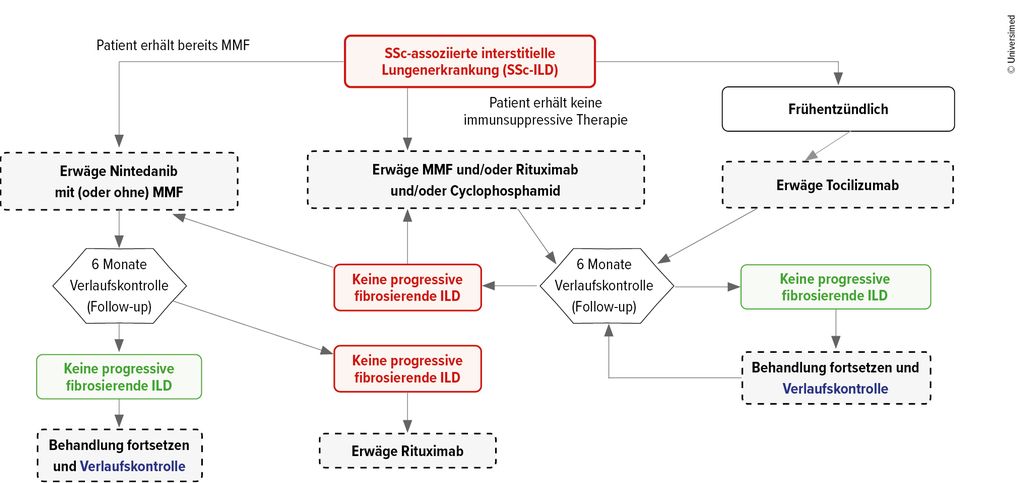
Abb. 2: EULAR/EUSTAR-Empfehlungen zur Behandlung der SSc-ILD
Argumente für eine frühe Intervention
Eine ausgereifte, über Jahre bestehende fibrotische Läsion sei in der idiopathischen Fibrose der Lunge (IPF) und wahrscheinlich auch anderer Organe weitgehend irreversibel. Ein massiv quervernetztes Bindegewebe, das kaum noch Zellen enthält, lasse sich auch nicht mehr wirklich entfernen oder mit den vorhandenen Therapieansätzen behandeln, erklärte Distler. Da die Lunge ein wenig regeneratives Organ sei, müsse man nach heutigem Stand davon ausgehen, dass verlorene Funktion und verlorenes Lungenvolumen in der Regel verloren bleiben, wenn fibrotische Ursachen zugrunde liegen.
Für eine frühe Behandlung spreche, dass moderne Therapieoptionen eine Verschlechterung verzögern oder vielleicht bestenfalls stoppen könnten. Doch bilde keiner der Ansätze fibrotische Veränderungen zurück. Das habe auch die SENSCIS-Studie gezeigt, in der mit Nintedanib behandelte Patientinnen und Patienten im Verlauf eines Jahres zwar weniger Lungenvolumen verloren als jene unter Placebo, es aber „eher nicht“ zum Progressionsstopp kam. Dies sehe man für die meisten derzeit verfügbaren Behandlungen.8
Subklinische ILD: Es bleibt eher nicht dabei
Eine monozentrische Studie aus Oslo hat beleuchtet, wie hoch die Prävalenz der subklinischen ILD ist und ob Betroffene ein Progressionsrisiko haben. In die Studie wurden Patientinnen und Patienten eingeschlossen, die an SSc, Anti-Synthetase-Syndrom (ASS) oder Mischkollagenosen (MCTD) litten. Als subklinisch galten Teilnehmende, deren Lungenvolumen im HRCT um weniger als 5% verändert war, und zwar bei normaler Lungenfunktion mit einer forcierten Vitalkapazität (FVC) über 80% und ohne respiratorische Symptome. Eine klinische ILD war definiert als Lungenbeteiligung im HRCT über 5%, alternativ unter 5% verbunden mit einer restriktiven Veränderung, also einer FVC unter 80%, und/oder respiratorischen Symptomen.4, 9–11
In der Osloer Kohorte von 525 CTD-Patientinnen und -Patienten mit Kollagenose litten 56% an SSc, 26% an MCTD und 18% an ASS. Die Klassifikation im CT unter Verwendung der genannten Definition ergab: Von ihnen zeigten 43% keinerlei Hinweise auf eine ILD und 44% Hinweise auf eine klinische ILD. Immerhin 13% hätten die Definition für eine subklinische IDL erfüllt, die meisten davon mit SSc (45%), gefolgt von MCTD (13%) und Mischkollagenosen und ASS (9%). Dies entspreche jeweils 10% der MCTD- und ASS-Betroffenen sowie 15% der Menschen mit SSc.
Während einer Nachbeobachtungsdauer von 4,5 Jahren ergab sich über alle Kohorten eine Progredienz von 24%. In der Gruppe mit subklinischer ILD waren es 38% gegenüber 51% bei Betroffenen mit etablierter ILD. Über ein Drittel der Teilnehmenden mit subklinischer IDL und – bis dahin vermutet – relativ unbedenklichen Läsionen war progredient und erfüllte über den Beobachtungszeitraum die Definition einer klinischen ILD. Dies spricht nach Distlers Ansicht angesichts der schlechten Prognose der progredienten ILD für frühe Interventionen – auch wenn die Diskussion nicht abgeschlossen sei und weitere Risikostratifizierungen noch fehlten.
Fortschritte: Marker für die Krankheitsaktivität
Bisher messe man die Krankheitsaktivität bei Fibrosen nur indirekt als Anhäufung von Schaden über die Zeit. Die FAPI-PET-CT erlaube eine direkte Messung (FAPI: Fibroblast Activation Protein Inhibitor). Das Fibroblasten-Aktivierungsprotein alpha (FAP) exprimierten alle aktivierten Fibroblasten, sobald eine entzündliche Reaktion oder eine Fibrose vorliegt. Über die Bindung an den 68GA-FAPI-04-PET-CT-Tracer lasse es sich detektieren und man könne so Umbaureaktionen quantifizieren. Eine hohe Akkumulation des Tracers zu Baseline sei mit einem hohen Risiko für eine Progredienz verbunden gewesen, fehlende wesentliche Akkumulation nicht. So lasse sich die Krankheitsaktivität einschätzen und potenziell auch die Therapie steuern, erklärte Distler.12
Was EULAR und EUSTAR empfehlen
Besteht eine SSc-ILD, könne generell Nintedanib mit oder ohne Mycophenolat-Mofetil (MMF) gegeben werden, alternativ MMF und/oder Rituximab (RTX) und/oder Cyclophosphamid oder als dritte Option Tocilizumab. Letzteres insbesondere dann, wenn die Einschlusskriterien der entsprechenden Studie erfüllt seien, konkret falls eine frühe Erkrankung mit entzündlichem Phänotyp vorliegt (CRP erhöht, Arthritis, Myositis).13
Diskutiert werde eine Reevaluation bereits nach 3 statt nach 6 Monaten, da bereits dann eine Progression eintreten könne. Ist das der Fall, sollte das Präparat gewechselt oder – meist präferiert – ein weiteres potenziell synergistisches hinzugenommen werden. Bei stabiler Erkrankung solle die Therapie beibehalten werden.
Die Evidenzlage zu den Empfehlungen
Zur Evidenz für diese Empfehlungen hätten die randomisierten kontrollierten Scleroderma Lung Studies 1 und 2 wesentlich beigetragen, berichtete Distler. In Studie 1 sei Cyclophosphamid bei SSc-ILD nach 12 Monaten moderat, aber signifikant wirksamer gewesen als Placebo: Das FVC sank um 1 vs. 2,6% des Ausgangsvolumens. In Studie 2 habe sich nach 24 Monaten unter Cyclophosphamid verglichen mit MMF kein signifikanter Unterschied in der FVC-Veränderung ergeben und damit ein indirekter Hinweis auf die Wirksamkeit von MMF, bei besserer Verträglichkeit.14,15
In einer placebokontrollierten Phase-III-Studie wurde die Wirksamkeit von Tocilizumab bei früher diffuser kutaner SSc geprüft. Hinsichtlich des primären Endpunkts Verdickung der Haut ergab sich im mRSS (Modified Rodnan Skin Score) eine tendenzielle, statistisch nicht signifikante Besserung unter Tocilizumab, formal ein negatives Ergebnis. Jedoch blieben die als sekundäres Studienziel gemessenen FVC-Werte unter Tocilizumab weitestgehend stabil, während sie im Placeboarm deutlich abfielen, mit einer hochsignifikanten Differenz in allen Subgruppen, bei ILD 240 Milliliter. Dies seien signifikante Effekte des in den USA zugelassenen Wirkstoffs, und zwar eher auf die Prävention einer IDL-Entstehung.16, 17
Wie sich in der bisher größten randomisierten kontrollierten Studie (SENSCIS) zu SSc zeigte, reduziert die Behandlung mit Nintedanib den FVC-Verlust um knapp 50% gegenüber Placebo.8
Zum therapeutischen Ziel B-Zellen gebe es eine sehr kleine, etwas umstrittene placebokontrollierte Studie aus Japan, die unter Rituximab einen ungewöhnlichen Abfall im mRSS zeigte. Jedoch belege die Studie auch eine signifikante Differenz in der FVC-Änderung und damit einen ersten Wirknachweis für Rituximab. Dies werde durch eine pneumologische Studie bestätigt, in der Rituximab vergleichbar wirksam war wie das gesichert aktive Cyclophosphamid, bei besserer Verträglichkeit.18, 19
Quelle:
„Fibrotische Erkrankungen der Lunge“, Vortrag von Prof. Dr. Jörg Distler, Düsseldorf, im Rahmen der Session „Fibrotische Erkrankungen – eine interdisziplinäre Herausforderung“ anlässlich des DGRh 2024
Literatur:
1 Wijsenbeek M et al.: N Engl J Med 2020; 383: 958-68 2 Nihtyanova SI et al.: Arthritis Rheumatol 2014; 66: 1625-35 3 Walker UA et al.: Ann Rheum Dis 2007; 66: 754-63
4 Hoffmann-Volt AM et al.: Am J Respir Crit Med Care 2019; 200: 1258-66 5 Steen VD et al.: Ann Rheum Dis 2007; 66(7): 940-4 6 Elhai M et al.: Ann Rheum Dis 2017; 76: 1897-1905 7 Hoffmann-Vold AM et al.: Lancet Rheumatol 2020; 2: PE71-E83 8 Distler O et al.: N Engl J Med 2019; 380: 2518-28 9 Hoffman-Vold AM et al.: Arthritis Rheumatol 2015; 67: 2205-15 10 Andersson H et al.: J Rheumatol 2016; 43: 1107-13 11 Reiseter S et al. Rheumatology (Oxford) 2018; 57: 255-62 12 Bergmann C et al.: Lancet Rheumatol 2021; 3: e185-194 13 del Galdo F et al.: Ann Rheum Dis 2024; doi: 10.1136/ard-2024-226430 14 Tashkin DP et al.: N Engl J Med 2006; 354(25): 2655-66 15 Tashkin DP et al.: Lancet Respir Med 2016; 4(9): 708-19 16 Khanna D et al.: Lancet Respir Med 2020; 8: 963-74 17 Denton CP et al.: Eur Respir J 2019; 54: RCT1883 18 Ebata S et al.: Lancet Rheumatol 2022; 4: e546-55 19 Maher T et al.: Lancet Respir Med 2023; 11(1): 45-54
Das könnte Sie auch interessieren:
Was gibt es Neues zur rheumatoiden Arthritis?
Unter den entzündlich-rheumatischen Erkrankungen ist die rheumatoide Arthritis (RA) jene mit der größten Zahl an zugelassenen Medikamenten. Damit sind aber bei Weitem nicht alle Probleme ...
Verdacht auf Riesenzellarteriitis: schnell ins spezialisierte Zentrum
Riesenzellarteriitis (GCA) und die Polymyalgiarheumatica (PMR) sind Großgefäßvaskulitiden (LVV), die vor allem im höheren Lebensalter auftreten. Insbesondere die GCA erfordert schnelles ...

Glukokortikoide: Chancen und Risiken
Glukokortikoide sind im Management entzündlich-rheumatischer Erkrankungen nach wie vor unverzichtbar. Ihrem gut durch Evidenz gesicherten Nutzen stehen – vor allem bei längerfristiger ...