PAH: Therapiestandard ist die initiale Kombinationstherapie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Mehr als zehn Medikamente stehen zur Behandlung der bekannten Signalwege bei pulmonal-arterieller Hypertonie zur Verfügung. Und die pharmakologischen Behandlungsoptionen nehmen mit den Erkenntnissen zu den Pathomechanismen weiter zu. Vielversprechend ist das Fusionsprotein Sotatercept und auch Imatinib könnte zukünftig eine Rolle spielen.
Die Outcomes bei pulmonal-arterieller Hypertonie (PAH) haben sich in den letzten 20 Jahren aufgrund der stetigen Weiterentwicklung der Behandlungsstrategien signifikant verbessert. Mit Endothelin-Rezeptor-Antagonisten, Phosphodiesterase-Typ-5-Inhibitoren, c-GMP-Stimulatoren, Prostacyclin-Analoga und IP-Rezeptor-Agonisten stehen mehr als 10 Medikamente zur Verfügung, die die bekannten Signalwege bei PAH adressieren (Abb. 1).1 Zu den Neuentwicklungen bei den bewährten Pharmakotherapien gehören vor allem inhalative oder orale Applikationsformen und Kombinationspräparate.
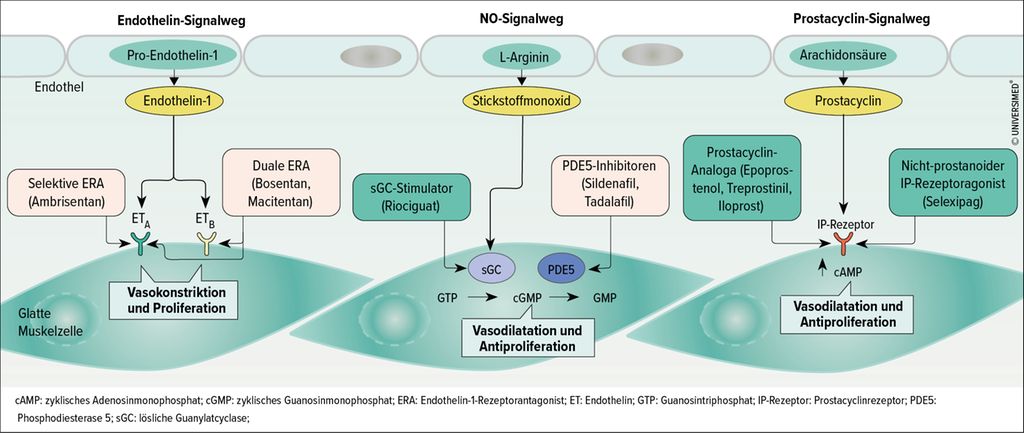
Abb. 1: Molekulare Therapieziele zugelassener Pharmakotherapien bei PAH (adaptiert nach Lau et al.)1
Initiale Dreifachtherapie: für wen?
Das Ziel der PAH-Behandlung ist ein niedriges bis moderates 1-Jahres-Mortalitätsrisiko (<5% resp. 5–10%).2 Internationale Guidelines empfehlen, Patienten mit neu diagnostizierter milder oder moderater PAH initial mit einer oralen Kombinationstherapie zu behandeln. Bei schwerer PH sollte die Therapie intensiviert werden (Abb. 2).3
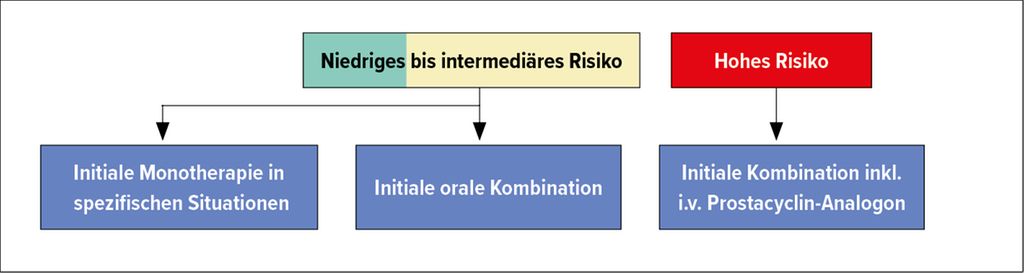
Abb. 2: Behandlungsempfehlungen bei neu diagnostizierter PAH (adaptiert nach Galié et al.)3
Welche Patienten von einer initialen Dreifachkombination am meisten profitieren würden, ist unklar. Zwei aktuelle Studien haben versucht, diese Frage zu beantworten. Die im letzten Jahr publizierte TRITON-Studie untersuchte den Effekt einer initialen Dreifachtherapie mit Macitentan, Tadalafil und Selexipag versus eine initiale Zweifachtherapie mit Macitentan und Tadalafil plus Placebo bei Patienten, die mehrheitlich an einer idiopathischen oder mit einer Bindegewebserkrankung assoziierten PAH erkrankt waren.4 Wie die Ergebnisse zeigten, führten die beiden Therapiestrategien zu einer vergleichbaren Reduktion der PVR («pulmonary vascular resistance»; primärer Endpunkt). Auch bezüglich der sekundären Endpunkte (6-MWD, NT-proBNP) zeigte sich kein Unterschied zwischen den verglichenen Therapien. Bei der zweiten Studie handelte es sich um eine retrospektive Analyse des französischen Patientenregisters für pulmonale Hypertonie. Diese hatte das 5-Jahres-Überleben in Abhängigkeit von der Therapie untersucht und gezeigt, dass von den Patienten, die initial mit einer Dreifachkombination behandelt worden waren, signifikant mehr am Leben waren als von denen unter einer initialen Zweifachkombination oder Monotherapie (91% versus 61% für die Zweifach-, resp. Monotherapie; p<0,001).5 Vor allem Patienten mit einem hohen oder intermediären Risiko profitierten von der initialen Dreifachtherapie, wie die Stratifizierung nach Risikogruppen zeigte.
Genetische Determinanten und Wachstumsfaktoren im Fokus
Fortschritte gibt es auch in Sachen Pathogenese: «In der Zwischenzeit haben wir gelernt, dass multiple Pathways zur Entstehung einer PAH führen können», so Dr. med. Mona Lichtblau vom Universitätsspital Zürich.
Einer der häufigsten genetischen Risikofaktoren für die Entwicklung einer hereditären und idiopathischen PAH ist eine Mutation des BMPR2(«bone morphogenetic protein receptor type 2»)-Gens. Diese verursacht eine Dysbalance von proliferativen und antiproliferativen vaskulären Wachstumsfaktoren. Die klinische Phase-II-Studie PULSAR hat die Wiederherstellung der pulmonalen vaskulären Homöostase mit dem Fusionsprotein Sotatercept (0,3oder 0,7mg/kgKG alle 3 Wochen s.c.) bei PAH-Patienten mit einer WHO-Funktionsklasse II–III und einer Backgroundtherapie mit einer Zweifach- oder Dreifachkombination untersucht.6 Wie die Ergebnisse der 24-wöchigen Studie zeigten, verbesserte sich die PVR (primärer Endpunkt) unter beiden Dosierungen Sotatercept signifikant im Vergleich zu Placebo. Auch hinsichtlich der sekundären Endpunkte (6-MWD, NT-proBNP) profitierten die beiden Verumgruppen deutlich. Die häufigsten unerwünschten Wirkungen unter der Behandlung mit Sotatercept waren ein Hämoglobinanstieg und eine Thrombozytopenie. Ein Patient in der Verumgruppe starb an den Folgen eines Herzstillstands. Sotatercept wurde ursprünglich zur Anämiebehandlung bei Patienten mit malignen hämatologischen oder hämolytischen Erkrankungen entwickelt. Aktuell wird das Fusionsprotein in einer Phase-III-Studie untersucht.
Ein weiterer vielversprechender Therapieansatz ist die Inhibierung von PDGF («platelet-derived growth factors») mit dem Tyrosinkinaseinhibitor Imatinib. 2013 hatten die Ergebnisse der IMPRES-Studie gezeigt, dass die PAH-Behandlung mit Imatinib hocheffektiv ist. Die Therapie wurde aufgrund schwerer unerwünschter Wirkungen, darunter das Auftreten von Subduralhämatomen bei Patienten, die gleichzeitig mit Antikoagulanzien behandelt wurden, nicht zugelassen.7,8 Aktuell wird Imatinib in inhalierbarer Form in einer Phase-II-Studie untersucht.
Körperliche Aktivität, Sauerstoff und Eisensupplementierung
Neben den pharmakologischen Behandlungsmöglichkeiten existieren verschiedene supplementäre Therapieansätze bei PAH. So können die Hämodynamik, Gehdistanz und Lebensqualität von PAH-Patienten durch ein spezialisiertes körperliches Training, das in der Rehabilitationsklinik begonnen und im häuslichen Setting fortgesetzt wird, verbessert werden.9 Eine Zunahme der körperlichen Leistungsfähigkeit und Lebensqualität konnte auch durch eine 5-wöchige Sauerstofftherapie erzielt werden, wie eine Studie am Universitätsspital Zürich zeigte.10 Keinen Einfluss auf die körperliche Leistungsfähigkeit und die Hämodynamik bei PAH zeigte in zwei kleinen Untersuchungen die Eisen-Supplementierung. «Diese Resultate stehen in Kontrast zu Studien bei Patienten mit chronischer Linksherzinsuffizienz, die eine Abnahme des Rehospitalisations- und Sterberisikos nach Eisensupplementierung zeigten», sagte die Referentin. Ob die Eisensupplementierung bei PAH wirklich obsolet ist oder ob das negative Ergebnis auf die unterpowerten Untersuchungen zurückzuführen ist, bleibt vorerst unklar.
Quelle:
Jahreskongress der Schweizerischen Gesellschaft für Pneumologie, 30. März bis 1. April 2022, Luzern
Literatur:
1 Lau EMT et al.: Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 2017; 14: 603-14 2 Galiè N et al.: 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37: 67-119 3 Galiè N et al.: Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801889 4 Chin KM et al.: Three- versus two-drug therapy for patients with newly diagnosed pulmonary arterial hypertension. J Am Coll Cardiol 2021; 78: 1393-1403 5 Boucly A et al.: Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am J Respir Crit Care Med 2021; 204: 842-54 6 Humbert M et al.: Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med 2021; 384: 1204-15 7 Hoeper MM et al.: Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013; 127: 1128-38 8 Kanaan R, Strange C. Use of multitarget tyrosine kinase inhibitors to attenuate platelet-derived growth factor signalling in lung disease. Eur Resp Rev 2017; 26: 170061 9 Grüning E et al.: Safety and efficacy of exercise training in various forms of pulmonary hypertension. Eur Respir J 2012; 40: 84-92 10 Ulrich S et al.: Effect of domiciliary oxygen therapy on exercise capacity and quality of life in patients with pulmonary arterial or chronic thromboembolic pulmonary hypertension: a randomised, placebo-controlled trial. Eur Respir J 2019; 54: 1900276
Das könnte Sie auch interessieren:


Ausgewählte Mitteilungen und Poster
Am Jahreskongress der Schweizerischen Gesellschaft für Pneumologie vom 15. bis 16. Mai 2025 in Genf gaben Schweizer Pneumologinnen und Pneumologen einen Einblick in ihre vielfältige ...

Biologika in der Asthmatherapie
Was ist zu beachten bei der Biologikatherapie für Menschen mit Asthma? Wann sollte sie sinnvollerweise begonnen werden und wie lange sollte sie fortgesetzt werden? Prof. Dr. med. ...