Molekularpathologie aggressiver B-Zell-Lymphome
Durch intensive Forschungstätigkeit der letzten Jahre ist es gelungen, die grosse Gruppe der diffusen grosszelligen B-Zell-Lymphome (DLBCL) in verschiedene Unterentitäten einzuteilen. Dies hat den Weg zu zukünftigen potenziell zielgerichteten Behandlungsoptionen eröffnet, gerade um deutlich aggressivere Verläufe vorhersagen und so adäquat therapieren zu können.
Keypoints
-
Schon in der aktuellen WHO-Klassifikation wurde versucht, Lymphome mit aggressivem Verlauf von den klassischen DLBCL abzugrenzen («High grade»-B-Zell-Lymphome).
-
Aktuelle sogenannte «Multi-Platform»-Studien an grossen Fallkollektiven zeigen die grosse molekulare Heterogenität der bisher als DLBCL, NOS klassifizierten Lymphome auf.
-
Weitere klinische Studien anhand dieser Algorithmen sind notwendig, um aufzuzeigen, ob sich die biologische Heterogenität auch in einem unterschiedlichen Ansprechen auf Therapien und eine unterschiedliche Prognose niederschlägt, um die Notwendigkeit einer Anpassung der pathologischen Klassifikationen zu belegen.
Die Änderungen der aktuellen WHO-Klassifikation 2016
Bereits in einer früheren Ausgabe der LEADING OPINIONS Hämatologie & Onkologie (2/2019) durften wir dieses Thema unter dem Blickwinkel der neuen WHO-Klassifikation präsentieren und möchten dies hier nur kurz zusammenfassen:1Neben der nun obligatorischen Ursprungszellklassifikation («cell of origin», COO) mit der Angabe, ob es sich um ein DLBCL mit Keimzentrums-Phänotyp (GCB-Typ) oder den Phänotyp der aktivierten B-Zelle (ABC-Typ) handelt, sollte auch der «double expressor score» bei DLBCL ermittelt werden, der die (immunhistochemisch bestimmte) Proteinexpression von c-MYC und BCL2 angibt. Diese prognostischen Angaben haben bisher keinen eindeutigen Einfluss auf die Therapie, es hat sich jedoch unter anderem gezeigt, dass Fälle mit einem hohen «double expressor score» unter R-CHOP schlechter verlaufen. Zudem wurde eine Gruppe besonders aggressiver Lymphome identifiziert, die sogenannten «high grade B-Zell-Lymphome (HGBL)». Diese sind entweder durch ihre blastäre, dem Burkitt-Lymphom ähnliche Morphologie oder das Vorliegen einer MYC-Translokation zusammen mit einer BCL2-/oder BCL6-Translokation oder auch allen drei Translokationen («Double hit/triple hit»-Lymphome) definiert. Dies ist ein erster Schritt der primär an Konsens und weltweit verfügbaren (und somit etwas eingeschränkten) diagnostischen Möglichkeiten orientierten WHO, die DLBCL-Kategorie zu stratifizieren.
Neue Forschungsergebnisse: Einblicke in die genetische Vielfalt des DLBCL
Bereits aufgrund der Morphologie wird die grosse Heterogenität der Entität DLBCLdeutlich (Abb. 1). Schon die COO-Klassifikation aus dem Jahr 2000 beruhte auf den Ergebnissen von Genexpressionsanalysen. Bei dieser Methode wird die Aktivität verschiedener Gene mittels Nachweis der jeweiligen RNA untersucht. Somit kann eine Herauf- oder Herunterregulation eines Gens nachgewiesen werden, und mittels komplexer statistischer Berechnungen können dann verschiedene Subgruppen gebildet werden, die bestimmte Eigenschaften teilen bzw. sich darin unterscheiden.
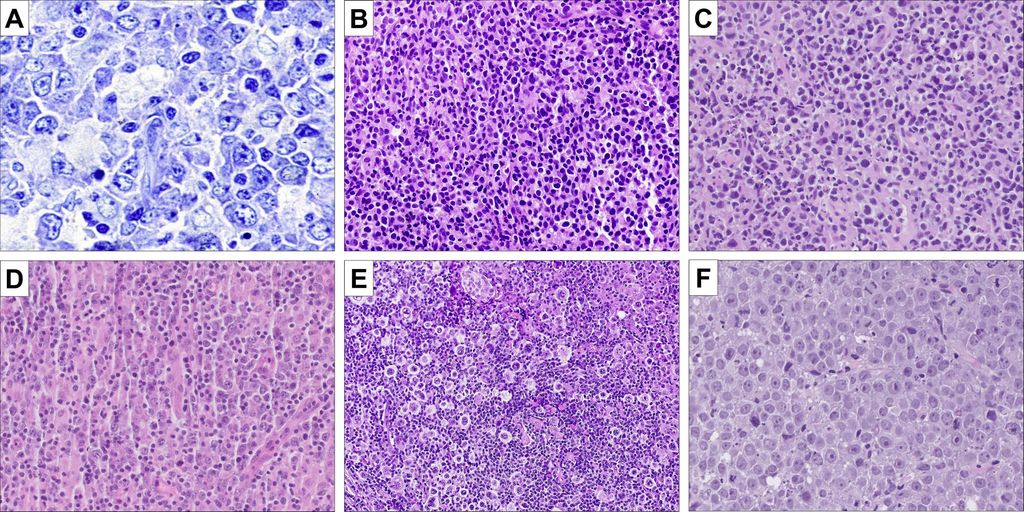
Abb. 1: Morphologisches Spektrum der DLBCL. A: DLBCL, NOS. In der Giemsa-Färbung zeigt sich die klassische Morphologie der Lymphomzellen (Giemsa, 400x); B: extranodales DLBCL des Magens mit diffuser Infiltration der Lymphomzellen vor einem Hintergrund aus nur kleinen reaktiven Lymphozyten (HE, 100x); C: DLBCL, CD5-positiv. Dieser Subtyp des DLBCL hat eine schlechtere Prognose. Morphologisch zeigen sich zahlreiche Apoptosen (HE, 100x); D: DLBCL, EBV-positiv. Dieser Fall zeigt ein deutliches reaktives Begleitinfiltrat aus Makrophagen und Lymphozyten (HE, 100x); E: seltener Fall eines anaplastischen DLBCL, welches zudem keine Expression von CD20 zeigte. Die deutlich atypischen Lymphomzellen liegen in einem dichten reaktiven Hintergrundinfiltrat aus Lymphozyten und Makrophagen; F: HGBL, NOS. Dieser Fall zeigt die klassische Morphologie aus blastoiden Zellen mit prominentem Nukleolus praktisch ohne reaktive Begleitinfiltrate (HE, 200x)
Wie in vielen anderen Bereichen der Genetik und Molekularbiologie ergaben sich auch bei Genexpressionsanalysen in den letzten Jahren mehrere methodische «Quantensprünge», was sich in neuen, sehr interessanten Forschungsergebnissen niederschlug: Die Studien von Sha et al.2 und Ennishi et al.3 konnten zeigen, dass sich auch in der Gruppe der nicht als HGBL definierten DLBCL ein signifikanter Anteil von Fällen mit einer molekularen Signatur von HGBL verbirgt und diese somit unterdiagnostiziert und auch untertherapiert sind, sofern man man die heute gebräuchlichen Methoden anwendet. So konnte auch die an sich sehr gute Prognose der GCB-DLBCL bestätigt werden, da die um die HGBLbereinigte GCB-DLBCL-Gruppe ein Gesamtüberleben von über 90% zeigte. Dies könnte auch ein Argument dafür sein, in dieser Patientengruppe eine Deeskalation der Therapie zu diskutieren, um eine Überbehandlung und somit unnötige Nebenwirkungen zu vermeiden.
Zwei weitere molekulargenetische «Multi-Platform»-Studien an jeweils mehrere Hundert Patienten mit DLBCL umfassenden Kollektiven zeigten auf, dass wahrscheinlich auch die COO-Einteilung zu simpel gedacht ist, um eine optimale prognostische Stratifizierung der Patienten zu erlauben: Schmitz et al. konnten die 574 Fälle ihrer Kohorte basierend auf den vorkommenden Mutationsprofilen und anderen genetischen Alterationen wie Änderung der Kopienzahl und Translokationen in fünf Gruppen mit unterschiedlicher Prognose einteilen,4 wobei auch die COO-Klassifikation sich schwerpunktmässig in verschiedenen Subgruppen abbilden liess. 55% der Fälle waren jedoch mit dieser Methode leider nicht klassifizierbar. Chapuy et al. untersuchten mit einem ähnlichen methodischen Ansatz 304 Patienten mit DLBCL, die ebenfalls in fünf genetische Gruppen eingeteilt wurden.5 Hier war der Anteil nicht klassifizierbarer Fälle deutlich geringer (12/304) und es zeigte sich, dass die fünf Gruppen die beiden COO-Gruppen in verschiedene Hochrisiko- und Niedrigrisikogruppen einteilen konnten. In beiden Studien zeigten insbesondere Fälle mit gleichzeitiger MYD88- und CD79B-Mutation («MCD») einen schlechten Verlauf und ähnliche Genexpressionsprofile.
Eine weitere Studie am erstgenannten Datensatz versuchte, der Tumorevolution gerecht zu werden.6 Mit der Annahme, dass ein Lymphom in seiner Entstehungmehrere Profile durchlaufen kann, wurde ein neuer Algorithmus namens «LymphGen» implementiert. Hierbei konnten neben den beschriebenen vier Subtypen drei weitere Subtypen unterschieden werden, die Anzahl der nichtklassifizierbaren Fälle ging auf 37% zurück. Auch die Fälle der anderen genannten Kohorte liessen sich mit «LymphGen» in diese 7 Subgruppen klassifizieren. Alle Subtypen wiesen klare Unterschiede bezüglich des Genexpressionsprofils, des Tumormikromilieus und auch der Prognose auf. Des Weiteren zeigte sich, dass bestimmte Subgruppen eine genetische Verwandtschaft zu niedrigmalignen B-Zell-Lymphomen aufwiesen, was darauf hindeutet, dass diese DLBCL nicht de novo,sondern als Transformation aus einem nicht klinisch in Erscheinung getretenen Vorläufer im Sinne eines niedrigmalignen B-Zell-Lymphoms entstanden sind.
Ausblick: von der Theorie in die Praxis
Diese Studien zeigen, dass sich das DLBCL in 7 genetische Subtypen unterteilen lässt. Allerdings wird auch der «LymphGen»-Algorithmus sicher nicht der letzte seiner Art sein. Gegenwärtig gilt eine wichtige Frage aber der Umsetzbarkeit dieser Ergebnisse im klinischen Alltag. Genexpressionsanalysen und auch umfassende Mutationsanalysen sind sicher nur an spezialisierten Zentren grossflächig einsetzbar und bedürfen neben der technischen Expertise auch Erfahrung im Umgang mit grossen Datenmengen.
Diese im Entstehen begriffenen Klassifikationen müssen sich erst im Alltag beweisen, wobei neben der allgemeinen Prognoseeinschätzung natürlich auch die Anwendung unterschiedlicher Therapieschemata eine Rolle spielt. Hierfür sind weitere prospektive Studien nötig; diese molekulare Klassifikation könnte aber die Voraussetzung für den längst fälligen und schmerzlich vermissten Fortschritt in der Primärtherapie des DLBCL darstellen.
Literatur:
1 Swerdlow SH et al.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition). Lyon, France: IARC; 2017 2 Sha C et al.: Molecular high-grade b-cell lymphoma: defining a poor-risk group that requires different approaches to therapy. J Clin Oncol 2019; 37(3): 202-12 3 Ennishi D et al.: Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. JClin Oncol 2019; 37(3): 190-201 4 Schmitz R et al.: Genetics and pathogenesis of diffuse large B-cell lymphoma. NEngl J Med 2018; 378(15): 1396-407 5 Chapuy B et al.: Molecular subtypes of diffuse large B-cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018; 24(5): 679-90 6 Wright GW et al.: Aprobabilistic classification tool for genetic subtypes of diffuse large B-cell lymphoma with therapeutic implications. Cancer Cell 2020; 37(4): 551-68 e1
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...