IPF und Lungenkrebs: doppeltes Risiko, halbe Optionen?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Lungenfibrose und Lungenkrebs haben gemeinsame Risikofaktoren. Daher treten beide relativ häufig gemeinsam auf, was die behandelnden Ärzt:innen vor besondere Herausforderungen stellt und die Prognose der Patient:innen deutlich verschlechtert.
Interstitielle Lungenerkrankungen (ILD), allen voran die idiopathische Lungenfibrose (IPF), sind vergleichsweise seltene Krankheiten, die durch ein variables Ausmaß an Inflammation und Fibrose zu einer meist irreversiblen Zerstörung der Lunge führen. Oft verlaufen ILD fortschreitend fibrosierend und haben dann eine sehr ungünstige Prognose, mit einem medianen Überleben von nur 3–5 Jahren für IPF.
Seit einigen Jahren sind mit den Substanzen Pirfenidon und Nintedanib sowie dem neuen und aktuell in der EU noch nicht zugelassenen Nerandomilast wirksame Medikamente für fortschreitende ILD verfügbar. Sie können den Verlust der Lungenfunktion bremsen und auch die Mortalitätsraten senken. Ein vollständiger Stopp der Erkrankung oder gar eine Regeneration von Lungengewebe ist aber weiterhin nicht möglich; die Prognose dieser Erkrankungen bleibt insgesamt ungünstig. Die Gesamtinzidenz der IPF als wahrscheinlich häufigste Form einer fortschreitend fibrotischen ILD liegt bei etwa 10/100000/Jahr. Deutlich häufiger ist sie bei Raucheranamnese, männlichem Geschlecht und höherem Lebensalter.
Abb. 1: Fallbeispiel: CT-Aufnahme mit neu aufgetretenem pulmonalem Plattenepithelkarzinom li. bei vorbekannter idiopathischer Lungenfibrose mit UIP-Muster, re. subpleural gut sichtbare Honigwabenzysten („honeycombing“)
Das Lungenkarzinom, vor allem in der nichtkleinzelligen Variante NSCLC, hat eine Inzidenz von 60/100000/Jahr und ist leider weiterhin die häufigste Krebstodesursache in Österreich. Trotz massiver Fortschritte in der Therapie, vor allem durch zielgerichtete, an Tumor-Genmutationen ausgerichteten Medikamenten und die breite Einführung von Checkpoint-Inhibitoren, liegt das mediane Überleben über alle Entitäten hinweg weiterhin nur bei 1–2 Jahren.
Lungenfibrose und Lungenkrebs haben gemeinsame Risikofaktoren, beispielsweise das Rauchen, gewisse Schadstoffexpositionen wie Asbest oder Silikate, männliches Geschlecht und höheres Lebensalter. Es ist also wenig überraschend, dass beide relativ oft gemeinsam vorkommen. Dies stellt Behandler:innen vor besondere Herausforderungen und verschlechtert die Prognose deutlich.
Wie häufig ist Lungenkrebs bei IPF?
Metaanalysen belegen, dass bei IPF-Patient:innen zwei Lungenkrebserkrankungen auf 100 Personenjahre auftreten. Dies ist etwa dreimal häufiger als im Lungenkrebsscreening-Setting bei Hochrisikopatient:innen mit älteren starken Rauchern (NLST-Studie; 0,6/100 Personenjahre). Ungefähr 10% der IPF-Patient:innen sterben am Lungenkrebs, was angesichts der hohen Mortalität der IPF selbst schon ein deutliches Signal ist. Der häufigste histologische Befund ist das Plattenepithelkarzinom, aber man geht davon aus, dass das Risiko für sämtliche Karzinome im Kontext einer IPF im Vergleich zu auf andere Riskofaktoren adjustierten Personen zumindest zweifach erhöht ist. Dies trifft besonders auch auf das kleinzellige Lungenkarzinom (SCLC) zu.
IPF und Lungenkrebstherapie
Es ist bekannt, dass das Vorhandensein einer IPF die Prognose von Lungenkarzinompatient:innen in allen Stadien signifikant verschlechtert. Nur für das SCLC konnte – wohl wegen der ohnehin sehr ungünstigen Prognose – kein derartiger Zusammenhang gezeigt werden. Die Gründe für diese Verschlechterung sind vielfältig:
-
Pulmonale Limitierung: Patient:innen mit IPF weisen oft schon früh im Krankheitsverlauf schwere Einschränkungen der Lungenfunktion, besonders des Gasaustausches, auf. Oft können diese Patient:innen trotz limitierter Stadien also nicht mehr kurativ operiert werden.
-
Selektion von Komorbiditäten: Sowohl IPF als auch Lungenkarzinom kommen meist nicht alleine. Durch die gemeinsamen Risikofaktoren beider Krankheiten haben viele Patient:innen auch kardiovaskuläre Komorbiditäten, begleitende COPD, neigen zur Kachexie und haben einen reduzierten Performancestatus.
-
Gehäufte Therapienebenwirkungen: Vor allem für Immuncheckpoint-Inhibitoren (ICI) ist eine höhere Rate von immuntherapievermittelter Pneumonitis bei ILD-Patient:innen bekannt. Obwohl diese Therapien auch bei ILD-Patient:innen ähnlich gut wirken würden, schließt die höhere Nebenwirkungsrate eine Erstlinientherapie mit ICI zulasten der Prognose oft aus. Auch verschiedene zytotoxische Chemotherapien oder „small molecules“ können, wenn auch seltener, zu Pneumonitis oder der Verschlechterung einer vorbestehenden ILD führen.
-
Akute Exazerbationen: Diese Ereignisse können bei fibrosierenden Lungenerkrankungen spontan oder auf externe Trigger wie Intubation/Beatmung, Lungenbiopsien, Operationen oder oft Infektionen auftreten und führen zu einem ARDS („acute respiratory distress syndrome“) mit oft unbeeinflussbar ungünstigem Verlauf. Es ist bekannt, dass diese Exazerbationen auch durch onkologische Therapien wie Lungenresektionen, Radio-, Chemo- oder Immuntherapien ausgelöst werden, was wohl zum Teil die erhöhte Mortalität von IPF-Patient:innen mit Lungenkrebs erklärt.
Was tun bei IPF plus Lungenkrebs?
Die Datenlage zu Therapieempfehlungen für Lungenkarzinome bei IPF-Patient:innen ist relativ dünn und beschränkt sich auf wenige, fast ausschließlich retrospektive japanische Arbeiten. Gezeigt werden konnte, dass Lungenoperationen mit einem deutlich erhöhten Risiko für akute Exazerbationen bzw. ARDS einhergehen, aber auch mit verlängerter Beatmungsdauer. Antifibrotische Therapien (Daten vor allem für Pirfenidon) könnten laut einigen kleinen Studien das operationsassoziierte Risiko vermindern. Dies impliziert erneut für die klinische Praxis, dass antifibrotische Therapien jedenfalls auch gegeben oder initiiert werden sollten, wenn ein Lungenkarzinom bei IPF auftritt.
Für die Radiotherapie ist das Komplikationsrisiko bei bestehender ILD ebenfalls deutlich erhöht: Retrospektive Arbeiten zeigten Pneumonitisraten zwischen 10 und 30%, auch mit gehäuft tödlichem Ausgang. Das Risiko ist von der Strahlendosis und vom Ausmaß des bestrahlten Lungengewebes abhängig.
Für Chemotherapien konnte gezeigt werden, dass (nab)Paclitaxel mit einer geringeren Komplikationsrate assoziiert sein könnte als Docetaxel oder Pemetrexed. Die Zugabe von Bevacizumab wurde als möglicher protektiver Faktor beschrieben, was insofern Sinn ergibt, als auch die Substanz Nintedanib als IPF-spezifisches Medikament VEGF hemmt. Die Kombination von Nintedanib mit Bevacizumab ist jedoch bis dato nicht erprobt und sollte in Anbetracht des wahrscheinlich erhöhten Blutungsrisikos unterlassen werden. Immuncheckpoint-Inhibitoren weisen schon per se ein gewisses Pneumonitisrisiko auf, besonders jedoch bei Menschen mit bestehender ILD, wobei die berichteten Inzidenzraten sehr schwanken (5–30%) – die Effektivität dieser Therapien scheint allerdings bei ILD-Patient:innen recht gut zu sein. Eine strenge Nutzen-Risiko-Abwägung ist daher jedenfalls erforderlich.
Die Tabelle zeigt die mögliche Herangehensweise bei Patient:innen mit NSCLC und bestehender ILD in Abhängigkeit vom Tumorstadium. Wichtig: Diese Auflistung reflektiert lediglich Expertenmeinungen und Ergebnisse kleiner, meist restrospektiver Studien. Ein individualisiertes und durch ein multidisziplinäres Tumorboard etabliertes Therapiekonzept ist unerlässlich!
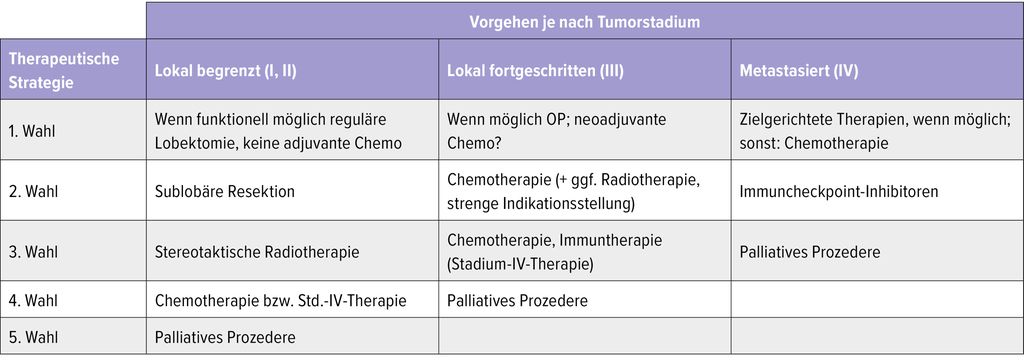
Tab. 1: Mögliche Herangehensweise bei Patient:innen mit nichtkleinzelligem Lungenkarzinom bei vorbestehender ILD, je nach Tumorstadium
Zusammenfassung
Interstitielle Lungenerkrankungen, vor allem die IPF, und Lungenkarzinome treten durch ähnliche Risikofaktoren (Rauchen, Alter, Geschlecht) gehäuft gemeinsam auf. Beide Krankheiten haben eine ungünstige Prognose, was sich in diesem Fall potenziert. Wichtig ist einerseits, die IPF indikationsgerecht mit antifibrotischen Therapien zu behandeln, da so eventuell das Krebsrisiko gesenkt werden kann. Andererseits ist auch bei bereits vorliegendem Lungenkarzinom eine antifibrotische Therapie sinnvoll, da dies anscheinend krebstherapiebedingte Komplikationen reduzieren kann. Die Therapieoptionen für Lungenkarzinome sind bei IPF-Patient:innen generell eingeschränkt, wichtig ist eine genaue multidisziplinäre Therapieentscheidung unter Einbeziehung sowohl des ILD-Boards als auch des Tumorboards.
Literatur:
beim Verfasser
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...