
Idiopathischer multizentrischer Morbus Castleman nach SARS-CoV-2-Impfung
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Der idiopathische multizentrische Morbus Castleman (iMCD) ist eine potenziell lebensbedrohliche, systemische, seltene Erkrankung mit komplexer Symptomatologie. Wir beschreiben das Auftreten eines iMCD mit TAFRO-Syndrom als Folge einer mRNA-Impfung gegen SARS-CoV-2 und die erfolgreiche Behandlung mit dem monoklonalen IL-6-Antikörper Siltuximab bei einem zuvor gesunden 20-jährigen Patienten.
Keypoints
-
SARS-CoV-2-Impfungen können auch seltene Nebenwirkungen haben, jedoch überwiegt bei Weitem der Vorteil der Impfung.
-
Wir konnten bei unserem Patienten zeigen, dass die Behandlung mit Siltuximab zu einer raschen und über sieben Monate nach Beendigung der Therapie anhaltenden kompletten Remission der Erkrankung führte.
-
Der vorgestellte Fall ist die Erstbeschreibung eines histologisch gesicherten iMCD nach mRNA-Impfung und könnte das Verständnis der Pathogenese der multisystemischen inflammatorischen Syndrome durch die Impfung als Auslöser ergänzen.
Klinischer Fallbericht
Ein 20-jähriger, bisher gesunder Student stellte sich in unserer Notaufnahme mit AZ-Verschlechterung und geschwollenen Lymphknoten vor. 18 Tage zuvor hatte er seine zweite mRNA-Impfung von Pfizer Biontech (Comirnaty) erhalten, vier Tage später traten erstmals hohes Fieber (39,5 Grad) und Atemwegsprobleme auf. In der Kindheit hatte er alle üblichen Kinderimpfungen bekommen und gut vertragen. Eine Allergieanamnese lag nicht vor.
In der klinischen Untersuchung fielen Anasarka, Petechien und generalisierte Lymphknotenschwellungen auf. Im Laborbefund waren u.a. die Thrombozyten (16000proμl) und die Leukozyten erniedrigt (2400proμl) und die Entzündungswerte wie PCT (2,6mg/dl), IL-6 (45,5pg/ml) und CRP (135mg/dl) teils deutlich erhöht. Eine vorherige oder derzeitige SARS-CoV-2-Infektion wurde ausgeschlossen.
In der Bildgebung mittels CT und MRT waren multiple vergrößerte Lymphknoten sichtbar, außerdem Aszites, Pleuraergüsse und eine Hepatosplenomegalie. Nach initialem Ausschluss autoimmuner, infektiöser und paraneoplastischer Ursachen entschieden wir uns zur Knochenmarkspunktion und Histologiegewinnung eines Lymphknotens.

Abb. 1: Histologische Aufarbeitung eines Lymphknotens mit regressiven Keimzentren (rGC) und Gefäßanomalien (Endothelien im Interfollikularraum HEV: „high endothelial vessels“)
Im Knochenmark fanden wir eine verstärkte Megakaryopoese und eine Fibrose von Grad 1. Ausgeschlossen werden konnten hämatologische Neoplasien und z.B. eine hämophagozytische Lymphohistiozytose (HLH). Im entnommenen Lymphknoten (Abb. 1) konnte eine M.-Castleman-typische Histologie auch mittels Referenzpathologie bestätigt werden. Auffällig waren eine polyklonale Plasmazellproliferation, atrophische Keimzentren und eine Proliferation der Endothelien in den Interfollikularraum. Es gab keine Hinweise auf eine HHV-8-Assoziation oder eine IgG4-assoziierte Erkrankung.
Therapeutisch war initial eine empirische Antibiotikatherapie mittels Amoxicillin/Clavulansäure gefolgt von Piperacillin/Tazobactam ohne klinischen Erfolg gegeben worden. Ebenso blieb eine hoch dosierte Kortisongabe über mehrere Tage ohne klinisches Ansprechen. Eine im April 2021 nach Diagnosestellung eingeleitete Therapie mit dem gegen IL-6 gerichteten monoklonalen Antikörper Siltuximab (11 mg/kg KG) führte zu einem raschen klinischen Ansprechen einhergehend mit einem Absinken der Entzündungsmarker (Abb. 2).
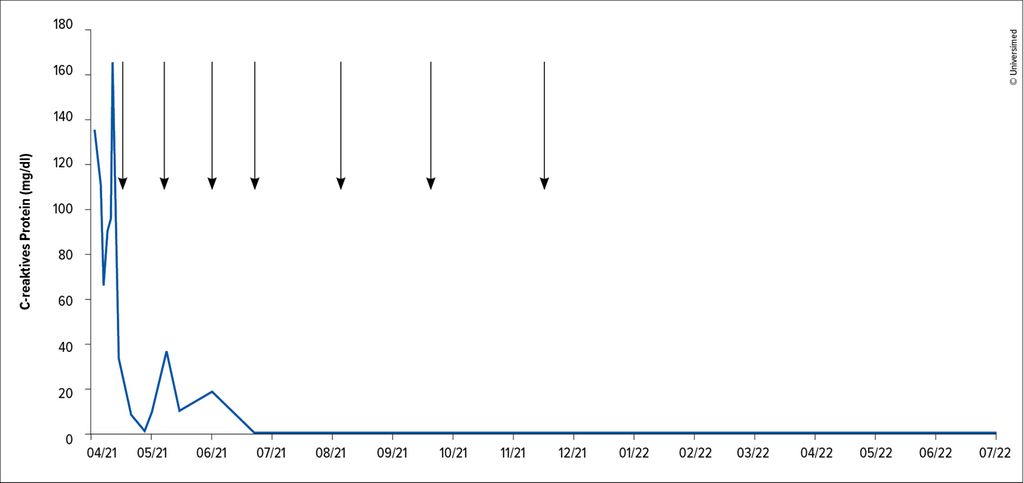
Abb. 2: Verlauf des C-reaktiven Proteins (blau, mg/dl) unter Siltuximab-Therapie (schwarze Pfeile)
Aus diesem Grund wurde die Therapie nach sieben Gaben beendet und über 18 Monate nach der Diagnose und sieben Monate nach der Therapie war der Patient frei von Symptomen ohne Anzeichen eines Rezidivs.
M. Castleman
Auch ca. 70 Jahre nach seiner Erstbeschreibung ist die Diagnosestellung und das Verständnis der Pathogenese des M. Castleman herausfordernd.1In unserer Veröffentlichung der Kasuistik werden die Erkrankung und die Pathogenese des iMCD ausführlich beschrieben und diskutiert.2 Unser Patient hatte zusätzlich ein TAFRO-Syndrom (Thrombozytopenie, Anasarka, Fieber, retikuläre Fibrose im Knochenmark, Organomegalie), welches auf einen schweren Verlauf hindeutet.
Für die Diagnosestellung des iMCD sind zwei große Kriterien (charakteristische Lymphknotenhistologie und multizentrische Lymphadenopathie) sowie mindestens zwei von elf kleinen Kriterien erforderlich. Unser Patient erfüllte sechs Kriterien (CRP-Erhöhung, Thrombozytopenie, Hypalbuminämie, Allgemeinsymptome, Hepatosplenomegalie und Pleuraergüsse). Obwohl die genaue Ätiologie des M. Castleman noch nicht vollständig bekannt ist, weiß man, dass IL-6 häufig zur Progression der Erkrankung beiträgt.3 Die einzige in Europa zugelassene Behandlung ist der Anti-IL-6-Antikörper Siltuximab.
Laut unserer Recherche ist unser Fall der weltweit erstein der Literatur beschriebene, der diese Nebenwirkung mit der mRNA-Impfung in Zusammenhang bringt und histologisch verifiziert ist. Auch wenn eine Kausalität schwer nachzuweisen ist, lassen das Ausbleiben eines Rezidivs und das rapide Ansprechen auf die Therapie einen temporären Trigger vermuten. Die aus den wenigen Daten bisher vermutete 5-Jahres-Überlebensrate bei iMCD mit TAFRO-Syndrom wird auf ca. 50% bis 75% geschätzt.2
Vor unserer Veröffentlichung war in Japan ein Patient einen Tag nach Comirnaty-Impfung an einem iMCD mit TAFRO-Syndrom erkrankt und verstorben,4 allerdings war hier die Diagnose nicht histologisch bestätigt worden. Autoptisch konnte der M. Castleman in den Lymphknoten nicht mehr nachgewiesen werden. Dieser Fall lässt vermuten, dass wir eventuell über das weltweit zweite vakzininduzierte Auftreten eines iMCD berichten.4
Systemische Inflammation
Bisher war für die Pathogenese einer systemischen Inflammation mit Zytokinsturm immer von vier bekannten Ursachen ausgegangen worden:5 neoplastische Ursachen, infektiöse wie z.B. virale Ursachen (z.B. HHV-8 oder EBV), iatrogene Ursachen (z.B. CAR-T-Zell-Therapie) oder autoimmune Ursachen (z.B. primäre oder sekundäre HLH).
Mit unserem Fall einer vakzininduzierten Auslösung eines idiopathischen multizentrischen Morbus Castleman könnten wir, insbesondere wenn durch weitere Fälle bestätigt, das Verständnis der Pathogenese dieser seltenen, aber schweren Erkrankung erweitern.
Literatur:
1 Van Rhee F: Nearly 70 years later: the continued unraveling of Castleman Disease. Haematologica 2022. Online verfügbar 2 Hoffmann C et al.: Idiopathic multicentric Castleman disease occurring shortly after mRNA SARS-CoV-2 vaccine. Vaccines 2022; 10(10): 1725 3 Van Rhee F et al.: International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018; 132: 2115-24 4 Yamada M et al.: TAFRO syndrome with a fatal clinical course following BNT162b2 mRNA (Pfizer-BioNTech) COVID-19 vaccination: a case report. J Infect Chemother 2022; 28(7): 1008-11 5Fajgenbaum DC, June CH: Cytokine storm. N Engl J Med 2020; 383(23): 2255-73
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...