
Hereditäre gastrointestinale Karzinome und ihre klinischen Implikationen
Gastrointestinale Tumoren, insbesondere Kolonkarzinom, Pankreaskarzinom und Magenkarzinom, gehören zu den führenden Krebstodesursachen. Ein relevanter Anteil dieser Erkrankungen hat einen genetischen Hintergrund. Deshalb ist das Wissen über mögliche vererbte Veranlagungen wichtig und die daraus folgenden präventiven und überwachenden Massnahmen sind essenziell.
Keypoints
-
Das Lynch-Syndrom hat verschiedene Risikoverteilungen für unterschiedliche Tumorerkrankungen. Die häufigsten sind Kolon- und Endometriumkarzinom, Ovarialkarzinom, Magenkarzinom, Urothelkarzinom und seltener auch Pankreaskarzinom.
-
Das HNPCC oder Lynch-Syndrom wird durch einen Funktionsverlust der DNA-Mismatch-Reparatur durch Mutation in den entsprechenden Genen verursacht.
-
Von Patienten mit Pankreaskarzinom haben ca. 10% eine positive Familienanamnese, nur etwa 10–15% davon sind bekannte Mutationen.
-
BRCA-Mutationen beim Pankreaskarzinom haben therapeutische Implikationen. Sie sprechen besser auf platinhaltige Chemotherapien an und es gibt Daten für eine Erhaltungstherapie mit Olaparib nach erfolgreicher platinhaltiger Erstlinienchemotherapie.
In diesem Artikel möchte ich näher auf das Kolonkarzinom und insbesondere auf das hereditäre nicht polypöse kolorektale Karzinom (HNPCC) bzw. Lynch-Syndrom eingehen, weiter auf das Pankreaskarzinom und die BRCA-Mutationen. Beide Syndrome sind unter den vererbten Veranlagungen für die beiden Erkrankungen jeweils die häufigsten und haben auch bei aktiver Tumorerkrankung therapeutische Implikationen, welche ich beleuchten möchte.
Hereditäres Kolonkarzinom
Bei Patienten mit Kolonkarzinom wird bei ca. 1–3% der Erkrankungen ein HNPCC-Syndrom gefunden.1 In der Erforschung dieser Tumorprädisposition war Henry Thompson Lynch massgeblich beteiligt, weshalb das Syndrom auch mit seinem Namen als Lynch-Syndrom bezeichnet wird. Henry T. Lynch hat zusammen mit weiteren Forschern anhand Familienbeschreibungen mit gehäuften Kolon- und Magenkarzinomen den vererbten Zusammenhang gesehen und phänomenologisch als Unterscheidung zu polypösen familiären Kolonkarzinomen die nicht polypöse Form beschrieben. Ein autosomal-dominanter Erbgang wurde festgestellt und weitere klinische Charakteristika ergänzt wie das Risiko für Tumoren des oberen Gastrointestinaltrakts, für Endometrium- oder Urothelkarzinome. Dies alles, bevor 1993 erstmals die genetische zugrunde liegende Ursache im Defekt der DNA-Mismatch-Reparaturgene gefunden wurde.2
Die DNA-Mismatch-Proteine sind bei der DNA-Reparatur dafür zuständig, falsch gepaarte Basen zu erkennen und auszuschneiden, sodass die Basen wieder korrekt gepaart werden können. Wenn dieser Mechanismus durch Gendefekte in den für diese Proteine codierenden Genen gestört ist, entstehen Fehler in der DNA-Replikation. Dadurch kommt es zur Anhäufung von Fehlern und konsekutiv auch zur Krebsentstehung. Sichtbar wird dies insbesondere durch die sogenannte Mikrosatelliteninstabilität.3
Personen mit einem Lynch-Syndrom haben ein deutlich erhöhtes Lebenszeitrisiko für Kolonkarzinome, Endometrium- und Ovarialkarzinome, Karzinome des oberen Gastrointestinaltraktes und je nach Familiengeschichte auch für Urothelkarzinome und Pankreaskarzinome. Aber auch andere Tumorerkrankungen wie zum Beispiel das Mammakarzinom oder Hauttumoren treten häufiger auf. Je nach zugrunde liegender Mutation beträgt das Risiko, bis zum 70. Lebensjahr ein Kolonkarzinom zu entwickeln, bis zu 50% – wobei die Streubreite gross ist.
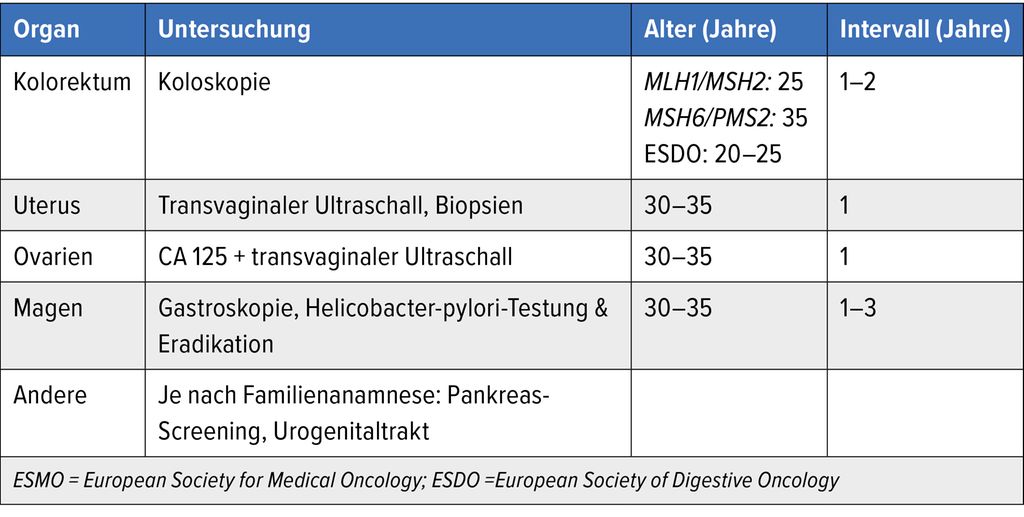
Tab. 1: Screening-Empfehlungen beim HNPCC (nach ESMO-Guidelines 2019 und ESDO-Empfehlungen 2018)
Das Risiko ist für Träger von Mutationen im MLH1- und MSH2-Gen am höchsten, im MSH6-Gen etwas weniger und am geringsten bei Trägern einer Mutation im PMS2-Gen. Daraus leiten sich gemäss internationalen Guidelines die entsprechenden Vorsorgeempfehlungen ab (Tab. 1).
Prophylaxe
Für das Kolonkarzinom gibt es Daten, dass eine prophylaktische Einnahme von Acetylsalicylsäure (Aspirin®) Tumorerkrankungen verhindern kann. Die Hemmung der Inflammation durch die Hemmung der Cyclooxygenase-2 und konsekutiv der Prostaglandin-E-Produktion soll diesen protektiven Effekt erklären. In der CAPP2-Studie wurde als Intervention für 2 Jahre Acetylsalicylsäure versus Placebo verglichen, wobei eine recht hohe tägliche Acetylsalicylsäure-Dosis von 600mg eingesetzt wurde. Es zeigte sich ein recht deutlicher Effekt für die Einnahme von Acetylsalicylsäure, allerdings wird der Effekt erst nach 4 Jahren sichtbar.4 Unklar ist, wie lange die Acetylsalicylsäure-Prophylaxe durchgeführt werden soll, und auch die Dosis ist unklar, da zur Hemmung der Cyclooxygenase-2 auch eine niedrigere Dosis ausreichen würde. Die Dosisfrage wird in der laufenden CAPP3-Studie untersucht, in der 600mg mit 300mg und 100mg verglichen werden. Aufgrund der jetzt vorhandenen Daten sollte eine Acetylsalicylsäure-Prophylaxe mit den betroffenen Personen vor dem Hintergrund des Blutungs- und Nebenwirkungsrisikos diskutiert werden.
Weiter ist es ausserordentlich wichtig, die Risikofaktoren Rauchen und Übergewicht zu adressieren.
Klinische Implikationen der Tumorgenetik
Wenn Patienten mit einem Lynch-Syndrom ein Kolonkarzinom entwickeln, hat dies in verschiedenen Stadien aus onkologischer therapeutischer Sicht Implikationen. So haben Patienten mit einem resezierten «Union for International Cancer Control (UICC)»-Stadium II, also Primärtumor ohne Lymphknotenbefall, eine insgesamt bessere Prognose als Patienten ohne Lynch-Syndrom und profitieren nicht von einer adjuvanten Chemotherapie.5 Im Stadium III ändert das Vorliegen eines Lynch-Syndroms das Vorgehen nicht, eine adjuvante Chemotherapie sollte den Patienten entsprechend den aktuellen Richtlinien empfohlen werden. Im Stadium IV ist die Therapie mit einem Immuncheckpoint-Inhibitor eine wichtige therapeutische Möglichkeit und seit der Publikation der Keynote-177-Studie auch als Erstlinientherapie einsetzbar.6
Beim Patienten mit einem Kolonkarzinom ist somit das Wissen über eine allfällige Mikrosatelliteninstabilität bzw. eine defiziente Mismatch-Reparatur essenziell, sodass bei diesen Patienten universell getestet werden soll. Als Initialtest kann gut die immunhistochemische Testung auf die Expression der DNA-Mismatch-Reparaturproteine erfolgen, da die Konkordanz zur Mikrosatelliteninstabilität mit ca. 98% sehr hoch liegt. Wichtig ist zu erwähnen, dass der grössere Teil der Mismatch-Defizienz bei den Kolonkarzinompatienten ein sporadisches im Tumor entstandenes Phänomen ist und nur wie eingangs erwähnt ein paar wenige Prozent der Patienten schliesslich auch wirklich ein zugrunde liegendes Lynch-Sydnrom haben. Bei Patienten mit einem MLH1/PMS2-Verlust sollten eine BRAF-Mutation und MLH1-Promotorhypermethylierung ausgeschlossen werden. Ein Tumor mit MLH1/PMS2-Defekt und vorliegender BRAF-Mutation kann als sporadisch betrachtet werden. Bei allen anderen Konstellationen sollte zusammen mit der Familienanamnese nach einem Lynch-Syndrom gesucht werden.7
Hereditäres Pankreaskarzinom
Von Patienten mit einem Pankreaskarzinom haben ca. 10% eine positive Familienanamnese und etwa bei 10–15% dieser Patienten findet sich eine genetische Tumorprädisposition, sodass die genetisch erklärbaren Pankreaskarzinome zum jetzigen Zeitpunkt eher selten sind. Die häufigsten zugrunde liegenden Vererbungen sind BRCA-Mutationen, «familial atypical multiple mole melanoma syndromes» (FAMMM), HNPCC, familiäre adenomatöse Polyposis (FAP), ATM-Mutationen, Peutz-Jeghers-Syndrom (PJS) und hereditäre Pankreatitis. PJS, hereditäre Pankreatitis und FAMMM haben die höchste Lebenszeitprävalenz. Mutationen im BRCA2-Gen werden am häufigsten gefunden.7
Vorsorgeuntersuchungen in Familien mit gehäuften Pankreaskarzinomen haben bisher keine sichere Verbesserung bzgl. Morbidität und Mortalität zeigen können. Dennoch wird gemäss dem Konsensus des «Consortium of the Pancreas Screening» (CAPS) Folgendes zum Screening empfohlen:8 MRT des Pankreas und Endosonografie als Baseline und danach im jährlichen Wechsel ab dem 40. Lebensjahr (PJS, hereditäre Pankreatitis und FAMMM) oder 45.–50. Lebensjahr (BRCA2, ATM, PALB2BRCA1, MLH1/MSH2), weiter ab dem 50. Lebensjahr für Personen mit familiärem Pankreaskarzinom (z.B. zwei oder mehr erstgradige Verwandte mit Pankreaskarzinom).
Therapeutische Implikationen entstehen unter anderem bei Patienten mit metastasiertem Pankreaskarzinom und Nachweis einer BRCA-Mutation. Hier konnte gezeigt werden, dass platinhaltige Chemotherapien ein besseres und längeres Ansprechen bringen können.9 Weiter wurde eine Verlängerung des progressionsfreien Überlebens mit einer Erhaltungstherapie mit dem PARP-Inhibitor Olaparib nach 16 Wochen erfolgreicher platinhaltiger Erstlinienchemotherapie gezeigt.10
Fazit
Abschliessend will ich nochmals auf die Wichtigkeit hinweisen, dass bei Patienten mit Tumorerkrankungen und auffälliger Familienanamnese oder auffälligen molekularen Analysen im Tumorgewebe diesen Befunden nachgegangen werden muss und genetische Beratungen in enger Zusammenarbeit mit Humangenetikern erfolgen sollen.
Literatur:
1 Lynch HT, de la Chapelle A: N Engl J Med 2003; 348(10): 919-32 2 Lynch PM: Revista Médica Clínica Las Condes 2017; 28(4): 500-11 3 Li GM: Cell Res 2008; 18(1): 85-98 4 Burn J et al.: Lancet 2020; 395(10240): 1855-63 5 Kim JE et al: Ann Surg Oncol 2015; 22(3): 630-37 6 André T et al.: N Engl J Med 2020; 383(23): 2207-18 7 ESMO Guidelines Committee: Ann Oncol 2019; 30(10): 1558-71 8 Goggins M et al.: Gut 2020; 69(1): 7-17 9 Golan T et al.: Br J Cancer 2014; 111(6): 1132-8 10 Golan T et al.: N Engl J Med 2019; 381(4): 317-27
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...