Diagnostische Algorithmen bei Leukaemia cutis
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Leukaemia cutis stellt eine spezifische Variante extramedullärer Leukämieerkrankungen dar. Neue Studien suggerieren, dass sie häufiger vorkommt als bisher gedacht. Im Folgenden soll ein Überblick über die Symptomatik, die histologische Abklärung und die Differenzialdiagnosen gegeben werden.
Leukaemia cutis (LC) beschreibt die Infiltration der Haut durch leukämische Zellen und stellt eine spezifische Variante extramedullärer Leukämieerkrankungen dar. Bei akuten myeloischen Leukämien (AML) und bei chronischen myeloischen Neoplasien (MN), insbesondere bei chronischer myelomonozytärer Leukämie, fallen solche Präsentationen in die Gruppe myeloider Sarkome. LC kann aber auch bei akuten lymphoblastischen Leukämien (ALL) auftreten.
Am häufigsten tritt LC im Rahmen bekannter (insbesondere rezidivierter) Leukämieerkrankungen auf, kann aber auch gleichzeitig (30% der Fälle) mit einer neu diagnostizierten Leukämie vorkommen, gelegentlich (<10% der Fälle) einer solchen vorausgehen oder ausnahmsweise die einzige Erkrankungspräsentation sein (die beiden Letzteren werden als aleukämische LC bezeichnet).1
Man geht davon aus, dass <5% der Leukämiepatient:innen von extramedullären Erkrankungen betroffen sind. Neuere Studien auf der Grundlage funktioneller Bildgebung (PET) implizieren allerdings, dass dies bei bis zu 20% der Patient:innen der Fall sein kann, und suggerieren eine unterschätzte Ausbreitungsneigung von AML, was eine erhebliche Morbiditäts- und Mortalitätsquelle sein kann.2,3
In einer umfassenden AML-Registerstudie an 1583 Patient:innen wurden 225 Fälle mit einer extramedullären Erkrankung festgestellt (ca. 15%), davon war eine Hautbeteiligung mit etwa 45% am häufigsten.4 Die Betroffenen wiesen höhere Leukozyten- und Blastenzahlen, monozytäre/-blastäre Differenzierung, PTPN11- & NPM1-Mutationen, FLT3-ITD und insbesondere die histologisch bestätigten Fälle verringerte Remissions- und erhöhte Frühsterblichkeitsraten und reduzierte Gesamt- und ereignisfreie Überlebensraten auf.
Die Überlebensunterschiede zwischen AML mit und ohne extramedulläre Manifestation schienen durch eine allogene hämatopoetische Zelltransplantation ausgeglichen zu werden,4 was darauf hindeutet, dass die immunologische Kontrolle über «Zufluchtsorte» ausserhalb des Knochenmarks eine Rolle bei der Verhinderung von Leukämierezidiven spielt. Besonders beachtenswert ist schliesslich, dass LC in der PET häufig negativ erscheinen, was den unübertroffenen Wert einer gründlichen klinischen Untersuchung unterstreicht.3
Symptomatik
LC-Patient:innen können einzelne oder mehrere Läsionen aufweisen, meist erythematöse oder hämorrhagische Papeln oder Plaques.5 Die am häufigsten betroffenen Körperteile sind die Beine, gefolgt von Armen, Rücken und Brust. Stellen, an denen zuvor oder gleichzeitig eine Entzündung aufgetreten ist, werden bevorzugt aufgesucht.
Klinisch kann die LC mit nichtneoplastischen kutanen Komplikationen von Leukämien verwechselt werden, die als «leukemids» bezeichnet werden und meist in Zusammenhang mit Zytopenien und Arzneimittelreaktionen stehen: Petechien, Purpura, Ekchymosen, leukozytoklastische Vaskulitiden, neutrophile Dermatosen, Herpesviruserkrankungen.5,6
Zusammen mit der o.g. Evidenz einer grösseren klinischen Bedeutung von nur histologisch bestätigten extramedullären Leukämiemanifestationen legt dies nahe, dass, wann immer möglich, eine histologische Bestätigung anzustreben ist.
Histologische Diagnose
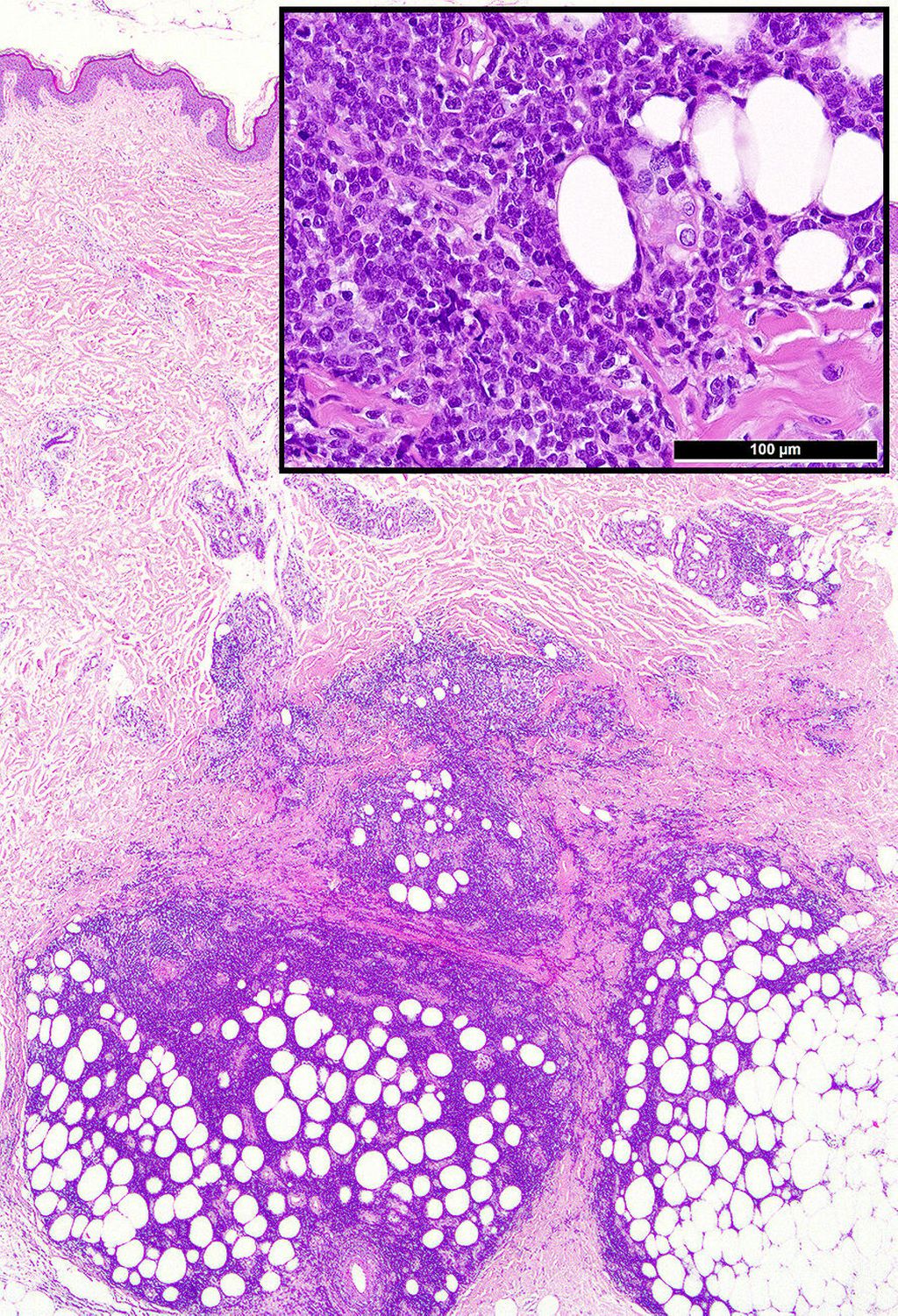
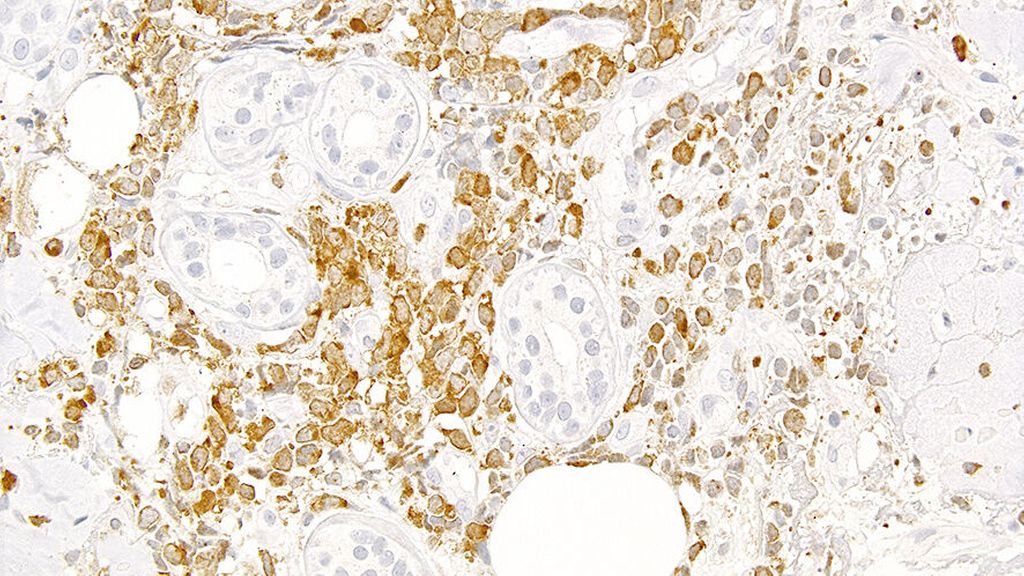
Abb. 1: Typische Präsentation einer Leukaemia cutis mit tiefenbetontem periadnexalem dermalem und interstitiellem bis kleinnodulärem subkutanem unreifzelligem (Einsatz-)Infiltrat
Die histologische Diagnose ist bei einer bekannten oder simultanen Leukämie einfach. Die meisten LC präsentieren sich als unreife klein-, blau- und rundzellige Tumoren (KBRZT) mit einer tiefer als erwarteten interstitiellen, perivaskulären, periadnexalen oder nodulären Infiltration der Dermis bzw. der Subkutis unter Aussparung der oberen Dermis (Grenzzone; Abb. 1); ALL betrifft in der Regel tiefere Strukturen.
Zu beachten sind mögliche abweichende phänotypische Besonderheiten (im Vergleich zum Knochenmark oder zum Blut) der blastären Hautpopulation, wie z.B. Expressionsverluste von CD34 und/oder CD117 und Expressionszugewinne von CD56, Lysozym und/oder Myeloperoxidase (MPO), welche auf bestimmte Subgruppen von Blasten, die bevorzugt extramedulläre Stellen besiedeln, zurückführbar sind.7
In klinisch kontextlosen Fällen sind erhebliche differenzialdiagnostische Schwierigkeiten zu erwarten, was auch dadurch unterstrichen wird, dass extramedulläre Leukämien zu den häufigsten Fehldiagnosen in der Pathologie gehören.8 Dies ist darauf zurückzuführen, dass bei KBRZT abgesehen von CD45, das schwach exprimiert oder negativ sein kann, selten Färbungen für hämatolymphoide Marker durchgeführt werden.
Differenzial- und Ausschlussdiagnosen
Bei Verdacht auf LC ist eine erste phänotypische Untersuchung mit CD43, CD45, INSM1, Lysozym, MPO, Pan-Zytokeratin (CK) und TdT ratsam, um den charakteristischen Phänotyp «CD43+, CD45±, INSM1–, Lysozym±, MPO±, CK–, TdT±» zu erheben. Bei abweichenden Kombinationen dieser Marker im Kontext von KBRZT mit Hautbeteiligung sind spezifische Zusatzuntersuchungen insbesondere zum Ausschluss von Merkelzellkarzinom, metastasiertem kleinzelligem Karzinom, synovialem oder Ewing-Sarkom, primitiven neuroektodermalen Tumoren etc. angezeigt.9
Wenn der beschriebene Phänotyp (CD43+, CD45±, INSM1–, Lysozym±, MPO±, CK–, TdT±) in einem kontextlosen KBRZT beobachtet wird, muss die Verdachtsdiagnose eines hämatolymphoiden Tumors ausgesprochen werden. Als Nächstes muss eine Neoplasie blastärer plasmazytoider dendritischer Zellen (BPDCN) durch zusätzliche Markeruntersuchungen für die charakteristische Konstellation aus Negativität für CD3, CD20, CD34, Lysozym und MPO und variabler Koexpression von CD4, CD56, CD68, CD123, CD303, IRF8, TCL1 und/oder TdT ausgeschlossen werden; CD303 wird als spezifisch angesehen.10
Nach diesem Ausschluss muss jeder LC-Fall der myeloischen oder lymphatischen Linie zugeordnet werden. Eine myeloide Differenzierung setzt eine CD3- und CD20-Negativität vor dem Hintergrund einer Positivität für CD14, CD68, CD117, CD123, IRF8, Lysozym und/oder MPO voraus, angenommen CD123 und IRF8 sind nicht koexprimiert, vermengte ortsständige reife plasmazytoide dendritische Zellen wurden in Betracht gezogen und hinsichtlich IRF8 liegt kein reifes B-Zell-Lymphom vor.
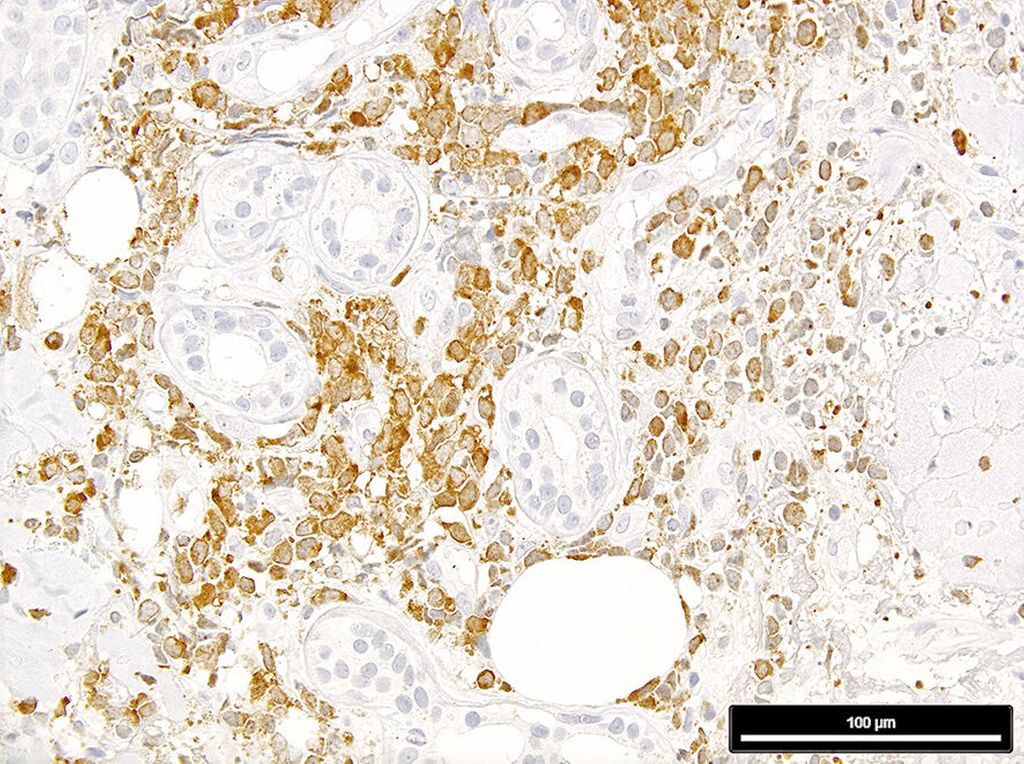
Abb. 2: Expression von mutiertem NPM1 in einer Leukaemia cutis (man beachte die negativen, da nichtmutierten, Hautadnexe); Immunperoxidase-Färbung mit dem mutationsspezifischen Antikörper PA1-46356
In einem letzten Schritt ist eine ICC2022/WHO-5-Diagnose zu stellen.11,12 Dafür sind zusätzliche Färbungen für p53 (eine starke diffuse Färbung indiziert eine AML/MN mit mutiertem TP53) und für NPM1 mit einem mutationsspezifischen Antikörper13 (eine zytoplasmabetonte Färbung indiziert eine AML/MN mit mutiertem NPM1; Abb. 2) sowie Analysen mit ausgewählten FISH-Sonden und NGS-Panels für subentitätsbestimmende genetische Aberrationen nötig.
In Fällen, welche negativ für myeloische Marker sind (schwache MPO-Expression bei B-ALL und CD117 bei früher T-Vorläufer-ALL [ETP-ALL] gestattet), erlaubt die Expression von entweder CD2, CD3, CD5, CD7, TCR-betaF1 und/oder -delta eine T-Zell-Linienzuordnung und von CD19, CD20, CD22, CD79a und/oder PAX5 eine B-Zell-Linienzuordnung.
In der T-Linie suggeriert die Expression von BCL11B, CD1a, CD10, CD34, CD117, LMO2, NOTCH1, TdT, TTF1 (Gen NKX2.1), PU.1 (Gen SPI1) einen unreifen Phänotyp. Gleiches gilt für CD10, CD34, CD99, ERG, schwach MPO, NUT1, SOX11, TdT im B-Linienkontext.14
In einem letzten Schritt ist eine ICC2022/WHO-5-Diagnose zu stellen.11,15 Die ETP-ALL ist phänotypisch definiert (CD1a–, CD2±, cCD3+, CD4±, CD5+, CD7+, CD8–, kombiniert mit der Expression von CD11b, CD13, CD33, CD34, CD65, CD117 und/oder HLA-DR), ETP-ALL mit BCL11B-Umlagerung phänotypisch und zytogenetisch, während alle anderen Fälle in die Kategorie T-ALL, NOS, fallen. Ausgenommen bei B-ALL mit BCR::ABL1- oder ETV6::RUNX1-Fusionen und MLL-, NUTM1- oder MYC-Umlagerungen ist eine subentitätsspezifische Diagnose mit den in den meisten Labors verfügbaren Diagnosemitteln nicht möglich, sodass einige Fälle notdürftig als B-ALL, NOS, bezeichnet bleiben werden.
Reifzellige nichtepidermotrope Lymphome mit Hautbefall sind sensu stricto keine LC, können aber (insbesondere blastoide Mantelzelllymphome, hochgradige B-Zell-Lymphome, T-Prolymphozytenleukämien, T-Zell-Lymphome im Panniculus usw.) differenzialdiagnostische Schwierigkeiten bereiten, die durch korrekte Anwendung diagnostischer Hilfsmittel bewältigt werden können. Ausführungen in dieser Hinsicht sprengen den Rahmen dieser Übersicht.
Fazit
Zusammenfassend stellt LC eine Differenzialdiagnose der KBRZT in der Haut dar. Ein – wie hier vorgeschlagen – schrittweiser phänotypisch-diagnostischer Ansatz, der die Schwächen und Stärken der verwendeten Marker berücksichtigt, kann in den meisten Fällen eine spezifische Diagnose ermöglichen.
Literatur:
1 Bakst R et al.: Diagnostic and therapeutic considerations for extramedullary leukemia. Curr Oncol Rep 2020; 22: 75 2 Buck AK et al.: First demonstration of leukemia imaging with the proliferation marker 18F-fluorodeoxythymidine. J Nucl Med 2008; 49: 1756-62 3 Stölzel F et al.: The prevalence of extramedullary acute myeloid leukemia detected by 18FDG-PET/CT: final results from the prospective PETAML trial. Haematologica 2020; 105: 1552-8 4 Eckardt JN et al.: Molecular profiling and clinical implications of patients with acute myeloid leukemia and extramedullary manifestations. J Hematol Oncol 2022; 15: 60 5 Wagner G et al.: Leukemia cutis – epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges 2012; 10: 27-36 6 Cho-Vega JH et al.: Leukemia cutis. Am J Clin Pathol 2008; 129: 130-42 7 Cronin DM et al.: An updated approach to the diagnosis of myeloid leukemia cutis. Am J Clin Pathol 2009; 132: 101-10 8 Solh M et al.: Extramedullary acute myelogenous leukemia. Blood Rev 2016; 30: 333-9 9 https://app.immunoquery.com/ 10 El Hussein S et al.: Plasmacytoid dendritic cells in the setting of myeloid neoplasms: diagnostic guide to challenging pathologic presentations. Br J Haematol 2023; 200: 545-55 11 Arber DA et al.: International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood 2022; 140: 1200-28 12 Khoury JD et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022; 36: 1703-19 13 Patel SS et al.: High NPM1 mutant allele burden at diagnosis correlates with minimal residual disease at first remission in de novo acute myeloid leukemia. Am J Hematol 2019; 94: 921-8 14 Duffield AS et al.: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 2023; 482: 11-26 15 Alaggio R et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022; 36: 1720-48
Das könnte Sie auch interessieren:
Erhaltungstherapie mit Atezolizumab nach adjuvanter Chemotherapie
Die zusätzliche adjuvante Gabe von Atezolizumab nach kompletter Resektion und adjuvanter Chemotherapie führte in der IMpower010-Studie zu einem signifikant verlängerten krankheitsfreien ...

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...