
Diagnostic visuel en endocrinologie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
En endocrinologie, le diagnostic repose généralement sur les valeurs de laboratoire. Certaines maladies peuvent néanmoins être diagnostiquées visuellement ou à l’aide d’examens cliniques simples. Les paragraphes suivants décrivent les signes cliniques particulièrement typiques de certaines maladies endocriniennes ainsi que la démarche diagnostique.
Syndrome de Cushing
Le syndrome de Cushing, également appelé hypercortisolisme, se caractérise par une répartition centrale et viscérale de la graisse, tandis que les extrémités restent minces, et une faiblesse musculaire proximale typique. Les caractéristiques typiques sont en outre des vergetures rouges (Fig.1), une peau parcheminée, une tendance aux hématomes, un visage «lunaire», une bosse prononcée au niveau de la nuque (bosse de bison) et une cicatrisation retardée des plaies. Les personnes concernées souffrent souvent d’obésité, d’hypertension artérielle et de (pré)diabète, d’humeur dépressive, d’œdèmes périphériques, d’acné, d’hirsutisme et d’anomalies du cycle menstruel. Les complications possibles comprennent les thromboses, les infections, l’ostéoporose, le développement d’un syndrome métabolique et une augmentation globale significative de la mortalité cardiovasculaire.
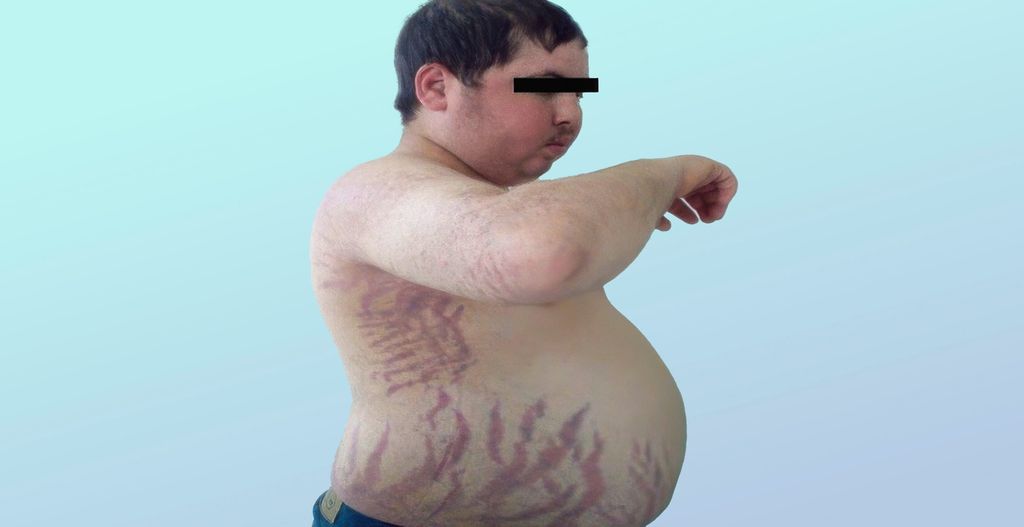
Fig.1: Caractéristiques cutanées typiques du syndrome de Cushing: vergetures rouges, peau parcheminée (source: El Aziz S et al.: Apport de l’orientation clinique dans le diagnostic des hypertensions artérielles endocriniennes. Pan African Medical Journal 2014; 18: 171. Sous licence Creative Commons Attribution 4.0 International [CC BY 4.0])
La cause la plus fréquente est un hypercortisolisme exogène, c’est-à-dire un hypercortisolisme indépendant de l’hormone corticotrope (ACTH), qui survient typiquement après un traitement de longue durée par des glucocorticoïdes. Le syndrome de Cushing endogène, beaucoup plus rare, résulte d’une augmen-tation endogène de la production de cortisol ou d’ACTH, par exemple en cas d’adénome hypophysaire ou surrénalien, ou encore dans le cadre d’un syndrome paranéoplasique.
L’examen diagnostique de référence est le test de freinage à la dexaméthasone. Dans ce cas, le patient prend 1mg de dexaméthasone à 23 heures. Si la cortisolémie est ≥50nmol/l le lendemain matin à 8heures, cela témoigne d’une sécrétion accrue de cortisol. D’autres analyses confirment le diagnostic, comme la mesure de l’excrétion de cortisol libre dans les urines de 24h ou du cortisol salivaire à minuit sur trois jours différents.
Le traitement dépend de la cause sous-jacente: les adénomes hypophysaires ou surrénaliens sont en premier lieu retirés chirurgicalement. De plus, la prophylaxie des thromboses et des infections est essentielle. Les options thérapeutiques médicamenteuses sont les inhibiteurs de la stéroïdogénèse comme le kétoconazole, la métyrapone ou l’osilodrostat.
Acromégalie
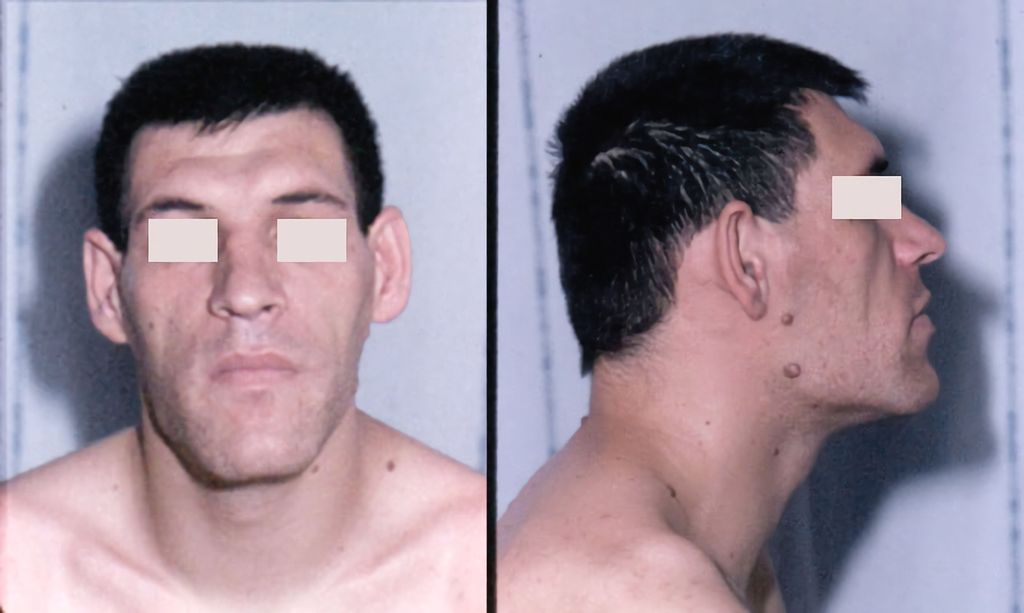
Fig.2: Traits du visage caractéristiques dans l’acromégalie (source: Chanson P, Salenave S: Acromegaly. Orphanet J Rare Dis 2008: 3; 17. Sous licence Creative Commons Attribution License 2.0 Generic [ https://creativecommons.org/licenses/by/2.0 ])
Les signes cliniques typiques de l’acromégalie ne surviennent souvent que tardivement, alors qu’il a déjà été possible d’observer une nette augmentation de la taille du visage avec croissance de la mâchoire inférieure (Fig.2), écartement des dents, macroglossie ainsi que, typiquement, une augmentation de la taille des mains (Fig.3) et des pieds. Toutefois, les personnes concernées souffrent généralement bien avant cela de symptômes tels que des céphalées, des douleurs articulaires, une transpiration excessive, un syndrome métabolique et, le cas échéant, un hypogonadisme. Un syndrome du canal carpien bilatéral à un jeune âge peut également ouvrir la voie à une suspicion clinique d’acromégalie.

Fig.3: Augmentation significative de la taille de la main dans l’acromégalie
Cette dernière est généralement causée par des adénomes hypophysaires qui entraînent une production excessive d’hormone de croissance (GH). Cela provoque une augmentation de la production d’IGF-1 («insulin-like growth factor 1») dans le foie, ce qui conduit à une croissance accrue des os, des muscles et des organes. En outre, une perturbation de la tolérance au glucose, un syndrome métabolique et des troubles mentaux peuvent survenir. Les personnes concernées présentent en outre un risque accru de polypes coliques et de cancer de l’intestin.
Le diagnostic est posé par la suppression insuffisante de l’hormone de croissance lors du test d’hyperglycémie provoqué par voie orale (75g de glucose). Le traitement consiste en premier lieu à retirer chirurgicalement l’adénome hypophysaire. Des médicaments tels que les analogues de la somatostatine (octréotide, pasiréotide), les antagonistes des récepteurs de l’hormone de croissance (pegvisomant) ou les agonistes dopaminergiques (cabergoline) peuvent être utilisés comme alternative ou en complément.
Maladie de Basedow

Fig.4: Goitre dans la maladie de Basedow (source: www.commons.wikimedia.com. Sous licence Creative Commons Attribution-Share Alike 3.0 Unported, 2.5 Generic, 2.0 Generic and 1.0 Generic)
Au premier coup d’œil, les patient·es atteint·es de la maladie de Basedow se distinguent par un goitre (Fig.4), une exophtalmie (Fig.5) et une rétraction palpébrale. Les yeux sont typiquement rouges et larmoyants, et les patient·es rapportent une diplopie. D’autres symptômes typiques de la maladie de Basedow sont des palpitations, une perte de poids, une transpiration excessive et, très caractéristiques, des troubles du sommeil. La maladie est déclenchée par des auto-anticorps anti-récepteur de la TSH (TRAK) qui stimulent la production d’hormones thyroïdiennes. Le diagnostic repose sur une baisse de la thyréotropine (TSH), associée à une augmentation de la thyroxine libre (fT4), de la triiodothyronine (fT3) et la présence de TRAK. Ces derniers présentent une sensibilité et une spécificité élevées pour la maladie de Basedow. De plus, l’hyperthyroïdie peut entraîner une élévation des transaminases et des phosphatases alcalines, une leucopénie et une anémie.

Fig.5: Orbitopathie dysthyroïdienne dans la maladie de Basedow
Le traitement de la maladie de Basedow consiste en des thyréostatiques (p.ex. carbimazole) pendant 12 à 18 mois. Le risque de récidive après l’arrêt du traitement est d’environ 50%. L’agranulocytose est un effet secondaire rare, mais grave, des thyréostatiques, raison pour laquelle un contrôle des leucocytes et des neutrophiles est effectué au début du traitement et, en particulier, en cas de thyréostatiques à forte dose. En cas d’orbitopathie dysthyroïdienne, il est recommandé de prendre des substituts lacrymaux et d’arrêter de fumer. Dans les cas sévères, un traitement par corticoïdes à forte dose ainsi qu’une décompression orbitaire chirurgicale peuvent être indiqués. En cas de récidive de la maladie de Basedow, un traitement par iode radioactif peut être envisagé, mais il risque d’aggraver une orbitopathie existante. Une thyroïdectomie peut également entrer en considération.
Hypothyroïdie
Les patient·es souffrant d’hypothyroïdie prononcée peuvent présenter un gonflement pâteux, en particulier au niveau périorbitaire (Fig.6). Dans les cas sévères, il·elles présentent une bradycardie et semblent apathiques. D’autres signes sont la sensibilité au froid, la prise de poids, la perte de cheveux et les anomalies du cycle menstruel ainsi que la diminution des réflexes. Le diagnostic se fait par mesure des taux de TSH et de fT4. En cas d’origine auto-immune, des taux élevés d’anticorps anti-thyroperoxydase (TPO) peuvent être détectés. Une hyponatrémie, une anémie et une hypercholestérolémie peuvent accompagner ces symptômes dans les cas sévères. Le coma myxœdémateux est une complication rare, mais sévère, avec un taux de mortalité pouvant atteindre 15%.
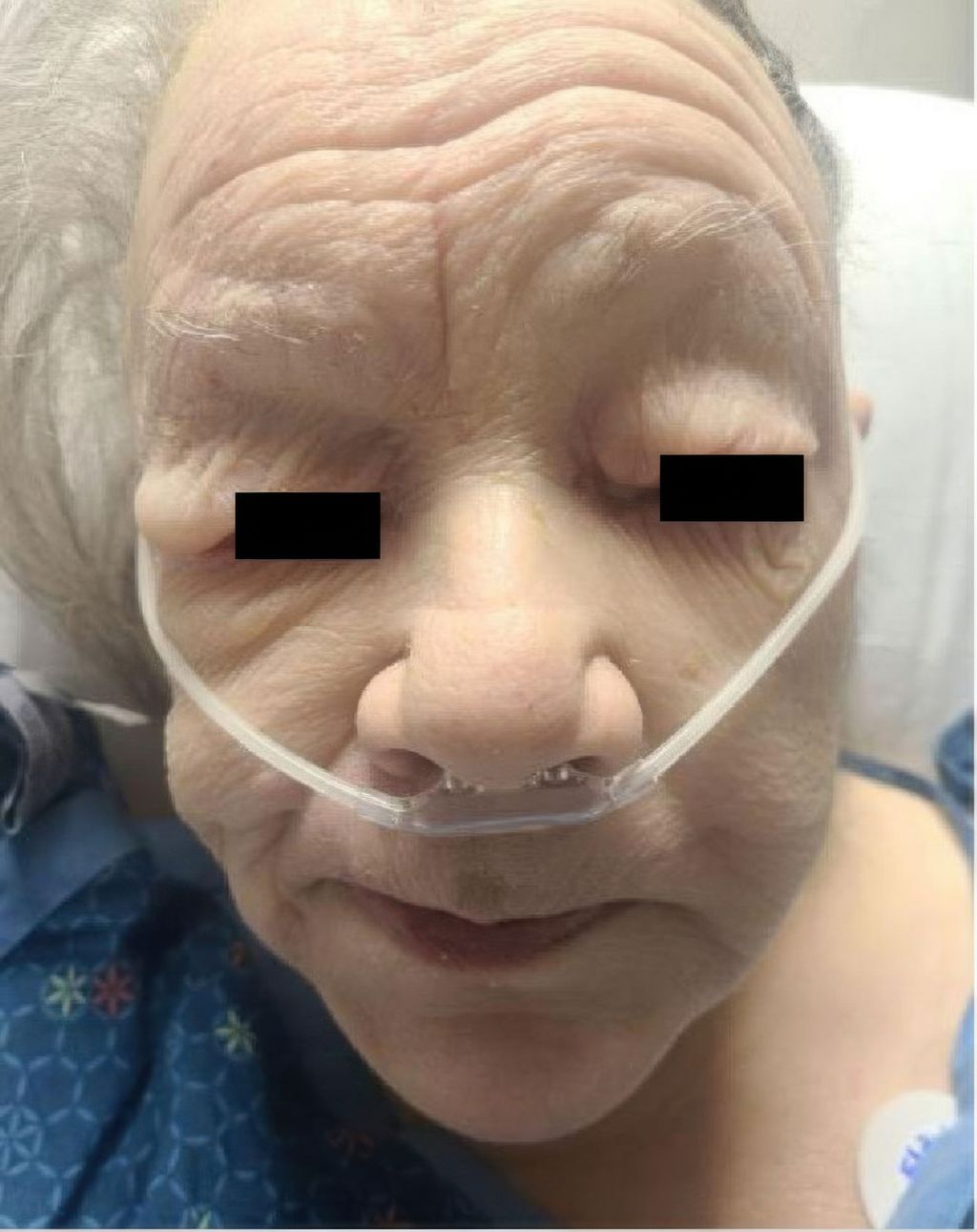
Fig.6: Myxœdème dans l’hypothyroïdie (source: Ponnapalli A et al.: Pericardial effusion uncovering underground hypothyroidism. Clin Case Rep 2021; 9: 1816-8. Sous licence Creative Commons Attribution License)
Le traitement repose sur la lévothyroxine à la dose cible de 1,6µg/kg/j, qui doit être augmentée progressivement chez les patient·es âgé·es ou ayant des antécédents cardiaques. En cas de myxœdème ou de suspicion d’autres déficits hormonaux, il faut d’abord exclure une insuffisance surrénale avant de prendre la lévothyroxine (300–500µg IV) afin d’éviter une crise addisonienne. En cas d’hypothyroïdie subclinique, il est recommandé d’initier le traitement à partir d’un taux de TSH >10mU/l.
En cas de maladie aiguë, par exemple dans un cadre hospitalier, les valeurs thyroïdiennes ne peuvent pas être interprétées avec certitude («non-thyroidal illness syndrome»).
Maladie d’Addison
L’insuffisance corticosurrénale primaire se manifeste notamment par une hyperpigmentation des plis palmaires (Fig.7), des cicatrices et de la muqueuse buccale. Celle-ci est due à la prohormone de l’ACTH (POMC). Les patient·es se plaignent d’une faiblesse marquée, d’adynamie, d’une perte de poids, d’hypotension. Une diminution des besoins en insuline dans le diabète de type 1 ou de nouvelles hypoglycémies peuvent également être des signes cliniques. Les modifications typiques des analyses de laboratoire sont des déséquilibres électrolytiques tels que l’hyponatrémie et l’hyperkaliémie.

Fig.7: Hyperpigmentation des plis palmaires dans la maladie d’Addison (source: Barts Health NHS Trust Archives SBHB/MU/14/27/9/2. Photo number: L0061738. Sous licence Creative Commons Attribution 4.0 International license)
L’une des causes possibles de la maladie d’Addison est la surrénalite auto-immune et, dans de plus rares cas, le carcinome corticosurrénalien, des métastases, des hémorragies, des maladies infectieuses (p.ex. tuberculose, infection à cytomégalovirus) ainsi que des maladies infiltrantes (p.ex. lymphome ou amylose). Une cortisolémie matinale <150nmol/l confirme le diagnostic d’insuffisance surrénale, tandis qu’une valeur >300nmol/l l’exclut. En cas de suspicion de maladie d’Addison, il convient d’initier rapidement une substitution par hydrocortisone afin d’éviter une crise addisonienne potentiellement fatale. Le traitement d’entretien consiste en une substitution par hydrocortisone (p.ex. 15–5–0mg), en plus de la prévention du stress et de l’éducation approfondie des patient·es. En cas de déficit concomitant en aldostérone, le traitement est complété par de la fludrocortisone.
Les signes cliniques en un coup d’œil
Syndrome de Cushing:faiblesse musculaire, vergetures rouges, hématomes, visage «lunaire»
Acromégalie:augmentation de la taille du visage/des extrémités, céphalées, transpiration, syndrome métabolique
Maladie de Basedow:orbitopathie dysthyroïdienne, palpitations, transpiration, troubles du sommeil
Hypothyroïdie:gonflement périorbitaire du visage, bradycardie, sensibilité au froid
Maladie d’Addison: hyperpigmentation de la peau, faiblesse, perte de poids, hypotension
Littérature:
● Fleseriu M et al.: Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol 2021; 9: 847-75 ● Nieman LK et al.: Treatment of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2015; 100: 2807-31 ● Fassnacht et al.: Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016; 175: 1-34 ●Fleseriu M et al.: A Pituitary Society update to acromegaly management guidelines. Pituitary 2020; 24: 1-13 ● Giustina A et al.: A Consensus on the diagnosis and treatment of acromegaly comorbidities: an update. J Clin Endocrinol Metab 2019; 105: dgz096 ● Bartalena I et al.: The 2021 European Group on Graves’ orbitopathy (EUGOGO) clinical practice guidelines for the medical management of Graves’ orbitopathy. Eur J Endocrinol 2021; 185: 43-67 ● Jonklaas J et al.: Evidence-based use of levothyroxine/liothyronine combinations in treating hypothyroidism: a consensus document. Thyroid 2021; 31: 156-82 ● Beuschlein F et al.: Glucocorticoid-induced adrenal insufficiency guideline resources. J Clin Endocrinol Metab 2024; 109: 1657-83 ● Vita JA et al.: Clinical clues to the cause of Addison’s disease. Am J Med 1985; 78: 461-6 ● Gravholt CH et al.: Clinical practice guidelines for the care of girls and women with Turner syndrome: Proceedings from the 2023 Aarhus International Turner Syndrome Meeting. Eur J Endocrinol 2024; 190: 53-151 ● Zitzmann M et al.: European Academy of Andrology guidelines on Klinefelter syndrome. Andrology 2020; 9: 145-67
Das könnte Sie auch interessieren:

Thérapies physiques en cas d’arthrite – nécessaires et utiles?
Le traitement des maladies articulaires rhumatismales inflammatoires a profondément évolué au cours des deux dernières décennies grâce à l’introduction de médicaments innovants: ...

Cancer du rein – stratégies actuelles et perspectives thérapeutiques futures
Les carcinomes rénaux non à cellules claires (non-ccRCC) sont pris en charge selon les mêmes standards que les carcinomes rénaux à cellules claires (ccRCC), mais les résultats ...

Chutes chez les personnes âgées: elles sont d’issue potentiellement fatale, mais aussi évitables
Les chutes chez les personnes âgées sont fréquentes et peuvent avoir de graves conséquences, mais elles ne sont souvent pas signalées par les personnes concernées en raison d’un ...