Lymphomatoide Papulose bei Kindern und Jugendlichen
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die lymphomatoide Papulose ist eine meist gutartig verlaufende, chronisch-rezidivierende Erkrankung, die histopathologisch maligne Lymphomerkrankungen imitieren kann. Diagnostisch ist die Korre-lation des klinischen Bildes und Verlaufes mit dem histopathologischen Befund von Bedeutung. Eltern betroffener Kinder sollten über die grundsätzlich gute Prognose, aber auch über das assoziierte erhöhte Malignomrisiko aufgeklärt werden.
Keypoints
-
Die LyP ist eine CD-30+ lymphoproliferative Erkrankung, die durch papulonoduläre oder ulzeröse, teilweise juckende Hautveränderungen gekennzeichnet ist, bei häufig chronisch-rezidivierendem Verlauf.
-
Die klinisch-histopathologische Korrelation spielt eine besondere Rolle bei der Diagnostik, da eine LyP klinisch mitunter nicht von anderen kutanen Lymphomerkrankungen unterschieden werden kann.
-
Aufgrund des selbstlimitierenden Charakters der LyP, der generell guten Prognose und des Fehlens langfristig kurativer Therapien kann eine Watch-and-wait-Strategie gewählt werden.
-
Je nach Ausprägung kann eine Therapie mit topischen Kortikosteroiden, NB-UVB oder niedrig dosiertem Methotrexat erfolgen. Antibiotische Therapie sollte nur bei superinfizierten Läsionen zum Einsatz kommen.
-
LyP bei Kindern und Jugend-lichen ist mit einer guten Prognose assoziiert. Das deutlich erhöhte Malignomrisiko erfordert jedoch ein lebenslanges hämatoonkologisches Screening.
Die lymphomatoide Papulose (LyP) ist eine seltene, chronisch-rezidivierende, CD-30+ T-Zell-lymphoproliferative Erkrankung, welche Erwachsene und seltener Kinder, Jugendliche und sogar Säuglinge betreffen kann.1 Buben sind etwas häufiger als Mädchen betroffen.1,2 Bedeutend ist die Diskrepanz zwischen dem meist gutartigen, klinischen Verlauf und dem scheinbar malignen histopathologischen Befund, welcher je nach Subtyp ein Hodgkin-Lymphom, eine Mycosis fungoides (MF) oder ein primär kutanes anaplastisch-großzelliges Lymphom (C-ALCL) imitieren kann.1,3 Die Ätiologie der LyP ist unbekannt. Viruserkrankungen wie das humane T-lymphotrope Virus 1, Herpes- oder Retroviren könnten möglicherweise eine Rolle spielen.1
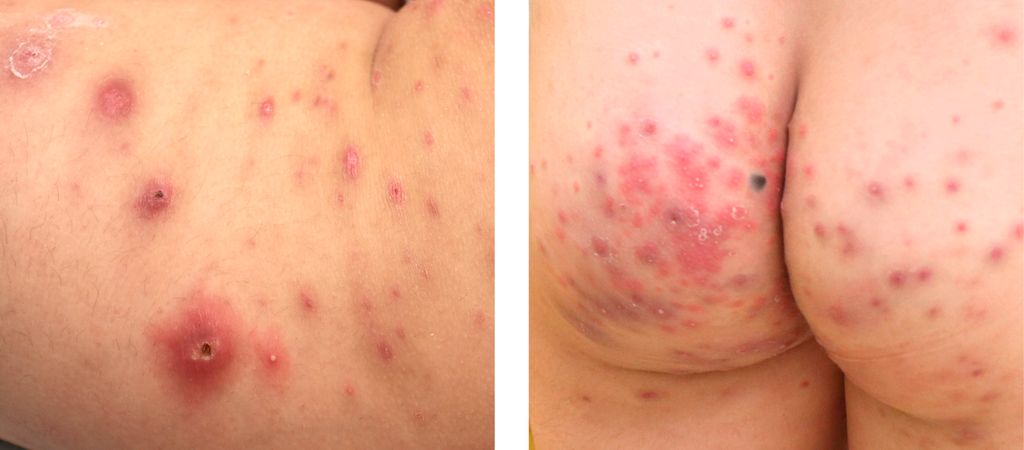
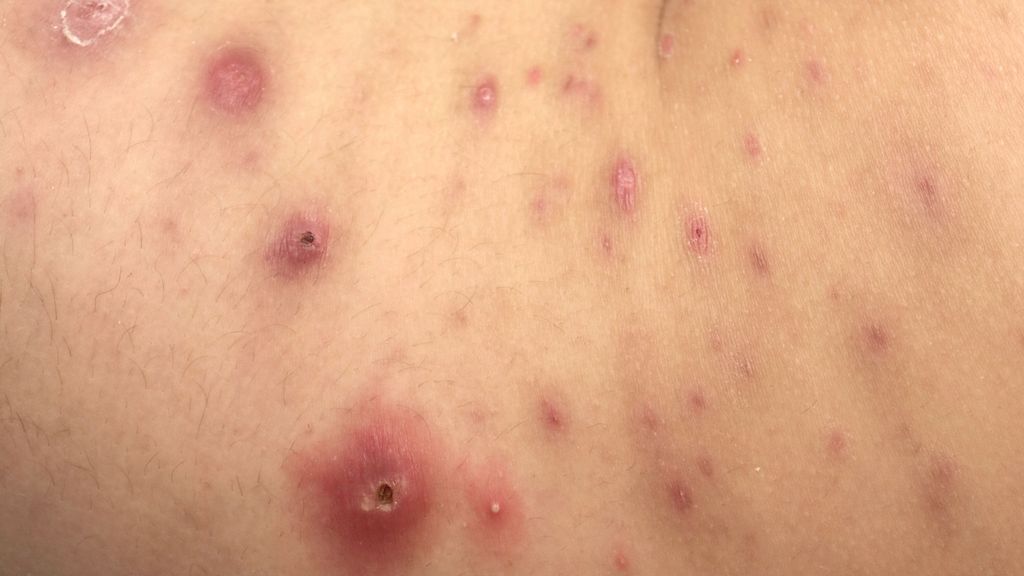
Abb. 1: Lymphomatoide Papulose: papulonekrotische Läsionen in unterschiedlichen Stadien

Abb. 2: Hämorrhagische Papeln, Crustae und Schuppung bei PLEVA
Hautstatus
Im klinischen Hautstatus finden sich erythematöse, papulonoduläre Hautveränderungen in verschiedenen Stadien, welche ein ulzeriertes oder nekrotisches Zentrum aufweisen können. Bevorzugt sind die Extremitäten befallen, am zweithäufigsten der Stamm. Die Läsionen können jedoch am gesamten Integument auftreten. Die LyP kann außerdem mit Juckreiz einhergehen, aber auch asymptomatisch sein.1,2
Klinischer Verlauf
Charakteristisch ist der selbstlimitierte, chronisch-rezidivierende Verlauf mit zwischenzeitlicher Abheilung und anschließend neuerlicher Eruption der Läsionen („waxing and waning“). Die Episoden dauern einige Wochen mit einem anschließenden, symptomfreien Intervall von einigen Monaten. Auch das Auftreten einer einzelnen Episode und einer kompletten Spontanremission ist möglich. Eine dauerhafte Krankheitslast ist in wenigen Fällen beschrieben.1 Nach Abheilung der Läsionen kann es zu Narbenbildung, Hyper- oder Hypopigmentierungen kommen.2
Diagnostik
Auf dem Weg zur richtigen Diagnose vergeht bei Kindern und Jugendlichen durchschnittlich mehr als ein Jahr.1 Aufgrund des Lokalbefundes kommt es in der Praxis häufig zu klinischen Fehldiagnosen wie Arthropodenreaktionen, Pityriasis lichenoides et varioliformis acuta (PLEVA), nodulärer Form einer chronischen Prurigo oder Follikulitis.1,2 Bei Verdacht auf eine LyP sollte eine Hautbiopsie mit immunhistochemischen Färbungen durchgeführt werden. Anhand des histopathologischen Bildes ist die Abgrenzung zu kutanen Lymphomerkrankungen, in erster Linie dem C-ALCL und einer großzellig transformierten MF, oft nicht möglich. Hier ermöglicht erst die klinisch-histopathologische Korrelation aus rezidivierenden, papulonekrotischen Hautveränderungen und den meist CD30+ atypischen Lymphozyten im histopathologischen Befund die Diagnose.1,2
Histopathologie
Eine LyP kann histopathologisch vielgestaltig auftreten und wird in 5 Subtypen (Tab.1) unterteilt, welche jeweils verschiedene Lymphome imitieren können. Am häufigsten tritt Subtyp A auf. Als Gemeinsamkeit aller histopathologischen Subtypen findet sich ein atypisches anaplastisches CD30+ lymphozytäres Infiltrat, in 5–10% der Fälle kann dieser Marker aber auch fehlen. Die Lymphozyten sind meist CD4+ oder CD8+ oder doppelpositiv für die beiden Marker. Doppelnegativität hingegen ist in nur wenigen Fällen beschrieben.1–3 Bei der LyP kann es außerdem zu einem Verlust der Zellmarker CD2, CD5 und CD7 kommen. In ungefähr zwei Dritteln der Fälle findet sich ein monoklonales T-Zell-Rezeptor-Rearrangement.4

Tab. 1: Histologische Subtypen und Differenzialdiagnosen der LyP (modifiziert nach Wieser I et al. 2016 und Willemze R et al. 2019)2,5
LyP versus PLEVA
Bei LyP zeigen sich Papeln und Noduli mit Krusten oder Nekrosen in verschiedenen Stadien, welche ohne Therapie heilen und meist rezidivieren. Bei PLEVA kommt es zu einem plötzlichen Auftreten von erythematösen Makeln und entzündlichen Papeln und in weiterer Folge zu Hämorrhagie, Verkrustung und Schuppung. Histopathologisch besteht bei LyP meist ein CD30+ lymphozytäres Infiltrat, bei PLEVA meist CD8+/CD30+ Lymphozyten.1 Die Differenzialdiagnose ist insbesondere bei CD30+ LyP oder CD30+ PLEVA erschwert und sollte dann anhand des klinischen Verlaufes erfolgen.1,6
Sekundäre Lymphome bei LyP
Kinder und Jugendliche mit LyP haben ein 87-fach erhöhtes Risiko, ein sekundäres Lymphom zu entwickeln, im Vergleich zu nicht betroffenen Gleichaltrigen. Wobei hier zu erwähnen ist, dass die Inzidenz hämatoonkologischer Erkrankungen bei Kindern sehr gering ist.1 In größeren pädiatrischen Kohorten wurde in 5–10% die Entwicklung eines hämatologischen Malignoms beobachtet.1,2 Risikofaktoren für Sekundärlymphome sind höheres Alter bei Erstmanifestation der LyP, männliches Geschlecht, häufige Episoden oder dauerhafte Symptomatik, T-Zell-Rezeptor-Monoklonalität sowie die histopathologischen Subtypen B und C. Bei den sekundären Lymphomen handelt es sich meist um eine MF oder ein ALCL, welche bei Kindern mit einer guten Prognose assoziiert sind. Sekundärlymphome können vor, während oder nach Erstmanifestation der LyP auftreten.1,3
Therapie
Watch and wait
Die derzeit verfügbare Therapie hat keinen Einfluss auf die Bildung sekundärer Lymphome und ist daher nicht zwingend erforderlich. Um Nebenwirkungen zu vermeiden, kann bei fehlendem Leidensdruck und nur einzelnen Läsionen eine Watch-and-wait-Strategie gewählt werden.1,2
Therapieoptionen
Bei quälendem Juckreiz oder hoher Krankheitslast mit Narbenbildung können mittelstarke bis starke topische oder intraläsionale Steroide verabreicht werden. Auch Narrowband-UVB und PUVA kommen zum Einsatz. Bei ausgeprägtem Befall ist auch der erfolgreiche Einsatz von niedrig dosiertem Methotrexat beschrieben. Antibiotische Therapie sollte nur bei Superinfektion der Läsionen zur Anwendung kommen. Präventiv kann ein antiseptisches Waschgel zum Duschen empfohlen werden.1,2
Prognose und Nachsorge
Grundsätzlich haben Kinder, die an einer LyP erkranken, eine gute Prognose mit einem 10-Jahres-Überleben von bis zu 100%. Des Weiteren kommt es bei Kindern im Gegensatz zu Erwachsenen seltener zu sekundären Lymphomen und häufiger zu einem milderen klinischen Verlauf mit Spontanremission.3
Lebenslanges Screening
In größeren pädiatrischen Kohorten wurde in 5–10% der Kinder und bis zu 22 Jahre nach Erstmanifestation der LyP das Auftreten von Sekundärlymphomen beobachtet. Daher wird ein jährliches Malignomscreening für mindestens 15 Jahre empfohlen.1,2 Es gibt diesbezüglich keine konkreteren Empfehlungen. Die Untersuchungen können den klinischen Status sowie eine Laborkontrolle inklusive Differenzialblutbild, gegebenenfalls eine Leukozytentypisierung, Organfunktionsparameter und eine Elektrophorese enthalten. Des Weiteren kann eine Sonografie der Lymphknotenstationen durchgeführt werden.
Literatur:
1 Blanchard M et al.: Paediatric-onset lymphomatoid papulosis: results of a multicentre retrospective cohort study on behalf of the EORTC Cutaneous Lymphoma Tumours Group (CLTG). Br J Dermatol 2024; 191(2): 233-42 2 Wieser I et al.: Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol 2016; 17(4): 319-27 3 Georgesen C, Magro C: Lymphomatoid papulosis in children and adolescents: a clinical and histopathologic retrospective cohort. Ann Diagn Pathol 2020; 46: 151486 4 Ceppi F et al.: Primary cutaneous lymphomas in children and adolescents. Pediatr Blood Cancer 2016; 63(11): 1886-94 5 Willemze R et al.: The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019; 133(16): 1703-14 6 Kempf W et al.: Paediatric cutaneous lymphomas: a review and comparison with adult counterparts. J Eur Acad Dermatol Venereol 2015; 29(9): 1696-709
Das könnte Sie auch interessieren:
Pityriasis lichenoides chronica
Die Pityriasis lichenoides chronica ist eine seltene, chronisch-entzündliche, papulosquamöse Hauterkrankung unklarer Genese mit schubweisem Verlauf. Trotz meist günstiger Prognose kann ...
Acne vulgaris – eine Nahrungsmittelallergie?
Allein die Fragestellung klingt verrückt, wo doch immer hormonelle Einflüsse als Ursache für Akne angeführt werden. Ziel dieses Beitrags ist es, die Hormone als alleinige Ursache infrage ...

Impfstoffe gegen humane Papillomviren
Humane Papillomviren sind weitverbreitete, teils sexuell übertragene Viren. Die Infektion verläuft in einer überwiegenden Zahl der Fälle symptomlos, kann jedoch gutartige Warzen sowie ...