.jpg)
Vom Routinefall zur Lebensgefahr: Lunge & Hirn – Duo infernale?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Abklärung unklarer pulmonaler Infiltrate gehört zum klinischen Alltag der Pneumologie und folgt meist einem infektiologischen oder onkologischen Algorithmus. Bisweilen kann aus einem Routinefall jedoch ein medizinisches Rätsel werden. Dieser Fall beschreibt die Odyssee einer Patientin, bei welcher erst der klinische Verlauf den Verdacht auf die seltene Diagnose einer Granulomatose mit Polyangiitis (GPA) lenkte.
Keypoints
-
Die Granulomatose mit Polyangiitis (GPA) bleibt eine diagnostische Herausforderung.
-
Dieser Fall unterstreicht die Bedeutung einer ganzheitlichen Betrachtung der Patient:innen und zeigt eindrucksvoll, wie in der Medizin der klinische Verlauf oft die Diagnose schreibt.
-
Wenn Lunge und Hirn als „Duo infernale“ auftreten, darf die Suche nach der gemeinsamen Ursache nicht an Fachgrenzen haltmachen.
-
Die Wachsamkeit gegenüber atypischen Verläufen und die Erweiterung des diagnostischen Fokus auf systemische Autoimmunerkrankungen sind entscheidend, um auch bei seltenen Entitäten wie der GPA rechtzeitig intervenieren zu können.
Ein Fall wie viele?
Im Oktober 2024 stellte sich eine 44-jährige Patientin (Hotelangestellte, aktiver Tabakkonsum mit kumulativ ca. 20–25 Packungsjahren) mit folgenden Beschwerden an einer pneumologischen Abteilung vor: Die Leitsymptomatik bestand aus akuten, atemabhängigen, rechtsseitigen Thoraxschmerzen mit Ausstrahlung in den rechten Arm sowie allgemeiner Abgeschlagenheit. Husten und Auswurf sowie eine klassische B-Symptomatik wurden verneint. In der Vorgeschichte war lediglich eine rezente Sinusitis maxillaris bekannt.
Die initiale klinische Untersuchung zeigte eine milde Sinustachykardie (HF 117/min) und eine leichte Hypertonie (165/96mmHg). Zudem präsentierte sich die Patientin subfebril (37,7°C), ansonsten waren der Status und die Vitalzeichen unauffällig. Das Routinelabor spiegelte eine mäßige systemische Inflammation wider: Leukozyten 12,6G/l, CRP 5,81 mg/dl. Wegweisend war jedoch die Bildgebung. In der konventionellen Röntgendiagnostik des Thorax zeigten sich zwei Konsolidierungen im rechten Oberfeld. Die anschließende Computertomografie offenbarte schließlich zwei markante Raumforderungen im rechten Oberlappen mit zentraler Einschmelzung. Unter dem Verdacht einer bakteriellen Genese wurde eine kalkulierte antiinfektive Therapie mit Ampicillin/Sulbactam eingeleitet.
Aufgrund steigender Entzündungswerte erfolgte eine Umstellung auf Piperacillin/Tazobactam mit zunächst gutem Ansprechen. Zudem wurde eine weiterführende Diagnostik mittels Bronchoskopie zur differenzialdiagnostischen Abklärung veranlasst. Trotz zwischenzeitlicher Besserung blieben mikrobiologische Untersuchungen inkl. auf Tbc (Blutkulturen, Sputum, bronchoalveoläre Lavage, mit endobronchialem Ultraschall gesteuerte transbronchiale Nadelaspiration; EBUS-TBNA) ergebnislos. Transbronchiale Kryobiopsien aus den Raumforderungen konnten aus dem anterioren und apikalen Oberlappensegment rechts gewonnen werden, deuteten jedoch lediglich auf eine organisierende Pneumonie mit fokalen Nekrosen ohne Malignitätsnachweis oder spezifische Erreger hin.
Das Duo infernale schlägt zu
Nach einer vorübergehenden klinischen Stabilisierung kippte das Bild dramatisch. Trotz breit angelegter Antibiose und mehrfacher Eskalation stiegen die Entzündungsparameter (CRP bis 30mg/dl) erneut an. Es entwickelten sich neue, alarmierende Symptome: Die Patientin zeigte klinisch eindrucksvoll einen rechtsseitig hängenden Mundwinkel sowie Sensibiliätsstörungen im Gesicht. Nach Ausschluss eines Apoplexes durch die Kolleg:innen der Neurologie wurde bei Verdacht auf eine ausgeprägte idiopathische periphere Fazialisparese rechts eine Steroidstoßtherapie empfohlen. Hierunter zeigte sich jedoch keine Besserung, vielmehr kam es in weiterer Folge zu einer zunehmenden klinischen Verschlechterung mit massiven hemikraniellen Kopfschmerzen und einer Hypoglossusparese rechts. Ein kraniales Kontrastmittel-CT lieferte den Verdacht auf eine ausgedehnte Phlegmone vom Pharynx bis zur Schädelbasis. Das „Duo infernale“ Lunge und Hirn war nun offensichtlich, doch die Ätiologie blieb ein Rätsel. Multifokale Abszesse ohne ausreichendes Ansprechen auf antiinfektive Maßnahmen mit zunehmenden Hirnnervenausfällen – nur eine vermeintliche Infektion?
Der Gamechanger
Die nun neue extrapulmonale Symptomatik leitete die diagnostische Kehrtwende ein. Die Kombination aus destruierenden Lungenprozessen, neurologischen Beschwerden und systemischer Inflammation führte zur erweiterten autoimmunologischen Abklärung. Die serologische Testung auf antineutrophile zytoplasmatische Antikörper (ANCA) zeigte eine hochgradige Positivität für Anti-Proteinase-3-Antikörper (27,0IU/mL [Fluoreszenz-Enzym-Immunoassay hochsensitiv; 0,0–3,0]) und brachte somit den entscheidenden Hinweis auf eine ursächliche Vaskulitis.
Zur weiteren Stadieneinteilung wurde ein 18F-FDG-PET/CT durchgeführt. Dieses wies eine hohe metabolische Aktivität auf – nicht nur in den pulmonalen Veränderungen, sondern auch im Bereich des raumfordernden Pseudotumors an der gesamten rechten Schädelbasis sowie in weiteren multifokalen Läsionen im Sinne eines generalisierten Stadiums mit Multiorganbefall (Abb.1).
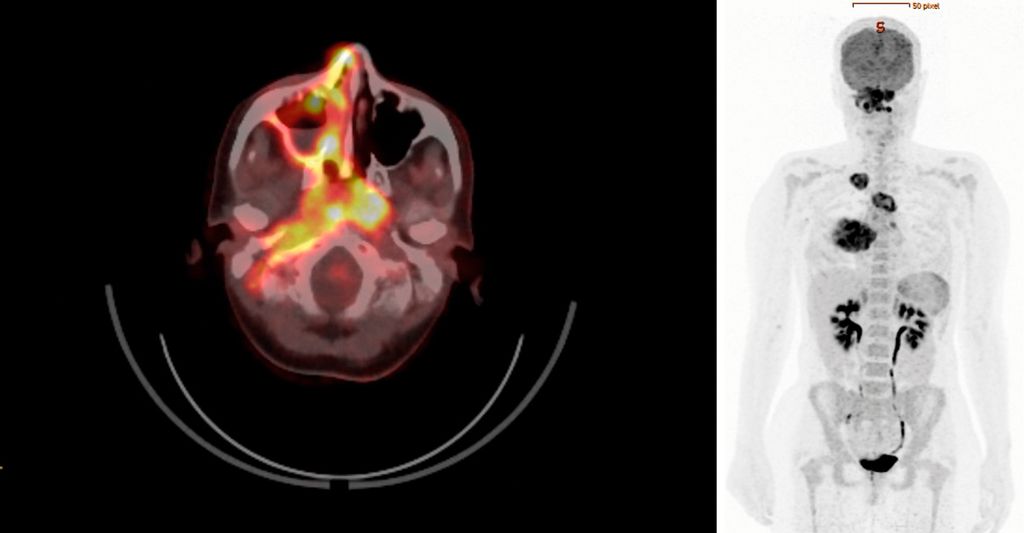
Abb. 1: 18F-FDG-PET/CT mit hoher multifokaler metabolischer Aktivität bei Vaskulitis mit Multiorganbefall (Quelle: Universitätsklinik für Nuklearmedizin Innsbruck)
Eine Reevaluation des mittels Kryobiopsie gewonnenen histopathologischen Probenmaterials aus den pulmonalen Raumforderungen und eine ergänzende endonasale Schleimhautbiopsie sicherten schließlich die Diagnose: eine Granulomatose mit Polyangiitis (GPA) mit pulmonaler und schwerer kranialer Beteiligung.
Multimodales Therapiemanagement und Verlauf
Die Situation der Patientin war zunehmend kritisch. Aufgrund progredienter neurologischer Ausfälle, insbesondere der Hirnnerven VII, VIII, IX, X und XII, bestanden u.a. eine ausgeprägte Dysphagie mit Aspirationsgefahr sowie eine rechtsseitige Stimmlippenparese. Das Management der Patientin wurde in der Folge interdisziplinär mit HNO/HSS, Neurologie, Neurochirurige, Thoraxchirurgie und Rheumatologie abgestimmt. Angesichts des organbedrohenden Verlaufs wurde eine intensivierte Remissionsinduktion eingeleitet. Das Therapiekonzept umfasste eine hoch dosierte Glukokortikoidstoßtherapie kombiniert mit zwei Zyklen Rituximab (RTX) 1000mg zur B-Zell-Depletion und zwei Zyklen Cyclophosphamid (CYC) 500mg als zusätzliche Eskalation aufgrund der Schwere der neurologischen Beteiligung. Außerdem erforderte der Zustand der Patientin ein interprofessionell koordiniertes Management inklusive Logopädie und Physiotherapie sowie die Anlage einer PEG-Sonde zur Sicherstellung der Ernährung bei schwerer Dysphagie.
Unter der eskalierten Immunsuppression kam es zu einer bemerkenswerten klinischen und radiologischen Besserung. Die Entzündungswerte normalisierten sich rasch und sowohl die pulmonalen Infiltrate (Abb.2) als auch der Pseudotumor an der Schädelbasis begannen in der Bildgebung zu schrumpfen. Auch die neurologischen Defizite zeigten sich rückläufig, wenngleich dieser Prozess deutlich langsamer verlief.

Abb. 2: Thorax-CT vor (links) und nach Einleitung der Therapie (Quelle: Universitätsklinik für Radiologie Innsbruck)
Ein Rückschlag im Verlauf und ein komplizierender Faktor war die Entwicklung einer sekundären hämophagozytischen Lymphohistiozytose (sHLH), vermutlich getriggert durch die massive Inflammation und Therapie, welche jedoch unter Anpassung des Regimes und engmaschigem Monitoring beherrscht werden konnte.
Mittlerweile befindet sich die Patientin in einer stabilen Remissionsphase mit anhaltend deutlich gebessertem Allgemeinzustand unter einer Erhaltungstherapie mit RTX und Mycophenolat-Mofetil (MMF), während die Glukokortikoide ausgeschlichen werden konnten. Die PEG-Sonde wurde wieder entfernt und die Patientin bereitet sich aktuell mit einer intensiven Rehabilitation zur Wiedereingliederung in den Arbeitsprozess vor.
Zusammenfassung: GPA als diagnostische Herausforderung
-
Vaskulitis als Chamäleon: Bei unklaren pulmonalen Einschmelzungen, die nicht auf eine antiinfektive Therapie ansprechen, muss die GPA immer differenzialdiagnostisch in Betracht gezogen werden – auch ohne klassische renale Beteiligung.
-
Extrapulmonale Wegweiser: Symptome im HNO-Bereich (Sinusitis, blutig-borkiger Schnupfen) oder neurologische Auffälligkeiten müssen bei Lungenerkrankungen als Warnsignal für eine Systemerkrankung verstanden werden.
-
Diagnostikmix: Die Kombination aus ANCA-Serologie, moderner Bildgebung (PET/CT) und Histologie führt zur Diagnose. Die Biopsie bleibt der Goldstandard zur Diagnosesicherung der GPA.
-
Multimodale Therapie und interdisziplinäre Zusammenarbeit: Schwere, komplexe Verläufe erfordern eine frühzeitige, intensivierte immunsuppressive Therapie, um irreversible Organschäden zu verhindern, und ein spezialisiertes interdisziplinäres Team, um die Betroffenen optimal zu versorgen.
-
Die gute alte Klinik: Der klinische Verlauf ist meist richtungsweisend und aussagekräftiger als initiale Einzelbefunde – der Verlauf schreibt die Diagnose!
Weiterführende Literatur:
● Odler B et al.: Diagnose und Therapie der Granulomatose mit Polyangiitis und mikroskopische Polyangiitis – 2023: Konsens-Empfehlungen der Österreichischen Gesellschaften für Nephrologie (ÖGN) & Rheumatologie (ÖGR). Wien Klin Wochenschr 2023; 135(5): 656-74
Das könnte Sie auch interessieren:
Rauchen und Mundgesundheit
Tabakkonsum schädigt die Mundgesundheit auf mehreren Ebenen. Besonders gut belegt sind die Assoziationen mit Parodontitis, Zahnverlust, gestörter Wundheilung, implantologischen ...
Immuntherapie: Krankheitsmodifikation bei allergischem Asthma
Allergisches Asthma ist die einzige Form von Asthma, für die mit der Immuntherapie eine kausale, krankheitsmodifizierende Therapie zur Verfügung steht, die in manchen Fällen sogar ...
