
Die zentrale Rolle der geringabundanten Bakterien im frühkindlichen Atemwegsmikrobiom
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Das Lungenmikrobiom spielt eine große Rolle in der Atemwegsgesundheit, vor allem bei Krankheiten wie der Mukoviszidose (CF). Das kürzlich entwickelte Softwaretool „raspir“ ermöglicht die Bestimmung fast aller Arten in einer bakteriellen Lebensgemeinschaft – Ergebnisse einer In-silico-Analyse von Metagenomdaten pädiatrischer CF-Patienten zeigen deren Bedeutung. Mithilfe dieses Tools können zukünftig wichtige Faktoren, die das gesamte Mikrobiom beeinflussen, wie zum Beispiel ein probiotischer Behandlungsansatz, genau untersucht werden.
Keypoints
-
Bisher konnten die in geringer Menge vorkommenden Bakterienarten in sequenzierbasierten Mikrobiomanalysen taxonomisch nicht entschlüsselt werden.
-
Das Software-Tool raspir reduziert das Methoden-assoziierte Störsignal und ermöglicht damit die Bestimmung fast aller Arten in einer bakteriellen Lebensgemeinschaft.
-
Die in geringer Menge in den Atemwegen vorkommenden Bakterien stabilisieren das Netzwerk des gesunden Lungenmikrobioms.
-
Probiotische Behandlungsansätze mit hoher Bakteriendiversität und niedriger Dominanz einzelner Bakterienarten könnten das unterentwickelte Bakteriennetzwerk der Atemwege bei chronischen Lungenerkrankungen stabilisieren.
In der kulturunabhängigen mikrobiellen Diagnostik wird die gesamte DNA einer Patientenprobe isoliert, fragmentiert, sequenziert, und die DNA-Fragmente werden gegen eine Datenbank mit bakteriellen Referenzgenomen kartiert. Das Ziel ist die Bestimmung der mikrobiellen Lebensgemeinschaft in einem Organ wie zum Beispiel in den Atemwegen. Die Herausforderung ist, dass viele Mikroben kurze genetische Sequenzen mit Mikroben innerhalb ihrer Lebensgemeinschaft und mit weiteren Einträgen in der Referenzdatenbank teilen. Fehlerhafte Kartierungen der biologischen DNA-Fragmente auf homologe Sequenzen in der Referenzdatenbank führen zum falschpositiven Nachweis von bakteriellen Spezies, die in Wirklichkeit in der Probe garnicht existieren. Um den Anteil dieser falschpositiven Treffer zu minimieren, legen die Auswertepipelines einen Grenzwert fest, um nur Aussagen über die mit Sicherheit vorhandenen hochabundanten Bakterien zu treffen, also den Bakterien, die kumulativ einen Anteil von 90–99% der Lebensgemeinschaft ausmachen. Jedoch gehen dabei wichtige Informationen über die geringabundanten Spezies verloren.
Das Mikrobiom des Menschen setzt sich aus wenigen hoch- und vielen geringabundanten Bakterienarten zusammen.1 Es ist daher davon auszugehen, dass die geringabundanten Bakterien den Großteil an genetischer Diversität und funktioneller Flexibilität des Mikrobioms bestimmen.2 Das Entfernen dieser Bakterienarten zu Beginn der Datenauswertung führt also dazu, dass wir ein unvollständiges Bild der mikrobiellen Lebensgemeinschaft im menschlichen Körper erhalten.
Entwicklung eines Softwaretools zur Bestimmung von Bakterien mit hoher und geringer Abundanz aus komplexen Metagenomproben
Vor Kurzem publizierten wir ein Softwaretool („rare species identifier“; raspir), das die Verteilung der Fragmentstücke entlang des Genoms bzw. die Genomorganisation der bakteriellen Spezies untersucht.3 Vergleicht man Bakterien der gleichen Art, so ist die Anordnung der Gene im Genom ähnlich. Da die DNA in der Tiefensequenzierung randomisiert vervielfältigt und sequenziert wird, müssten in den tatsächlich vorhandenen Bakterienarten DNA-Fragmente aus allen Teilen des bakteriellen Genoms nachzuweisen sein. Im Gegensatz dazu verteilen sich falschpositive Sequenzabschnitte nicht gleichmäßig über dasgesamte Genom. Dementsprechend lassen sich mit einem mathematischen Kniff hoch- und geringabundante Bakterienarten von falschpositiven Treffern wie folgt unterscheiden: Man berechnet die Distanz zwischen allen DNA-Fragmenten, die einem Referenzgenom zugeordnet werden, zerlegt das Positionssignal in seine Spektralkomponenten und vergleicht das Signal im Frequenzbereich mit einem theoretischen Referenzsignal, das eine ideale Gleichverteilung der DNA-Fragmente simuliert.3 Abbildung 1 erläutert das Prinzip.
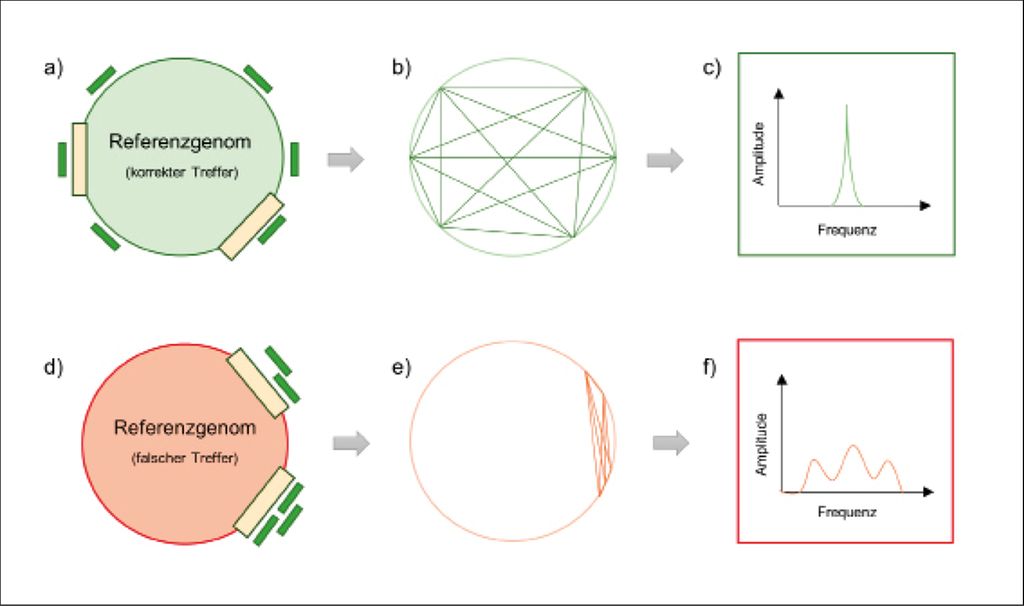
Abb. 1: Vereinfachte Darstellung, wie man die Platzierung von DNA-Stücken entlang eines Referenzgenoms nutzen kann, um das Vorkommen eines Bakteriums in der Patientenprobe nachzuweisen. (a) Wenn die biologischen DNA-Fragmente (grün) dem Genom des Bakteriums zugeordnet werden, das vorhanden war, verteilen sich die DNA-Stücke gleichmäßig über das Genom. (b) Die Distanz zwischen allen Fragmenten, die dem richtigen Referenzgenom zugeordnet werden, wird berechnet. (c) Das Positionssignal wird in seine Spektralkomponenten zerlegt und mit einem Referenzsignal verglichen, das diesem hier gezeigten Positionssignal ähnelt. (d) Wenn die DNA-Fragmente (grün) dem falschen Genom zugeordnet werden (rot), also einem Bakterium, das sich nicht in der Probe befand, aber genetische Sequenzen mit dem richtigen Bakterium teilt (gelbe Balken), häufen sich die Fragmentstücke an diesen Abschnitten. (e) Die Distanz zwischen allen Fragmenten wird berechnet. (f) Das Signal wird in seine Spektralkomponenten zerlegt und mit einem Referenzsignal verglichen, das dem Signal in Abbildung c ähnelt
Ergebnisse einer In-silico-Analyse von bereits publizierten Metagenomdaten
Um die Bedeutung der Bakterien mit geringer Abundanz in der frühkindlichen Entwicklung des Atemwegsmetagenoms bei gesunden und Kindern mit Mukoviszidose (CF) zwischen 0 und 6 Jahren zu analysieren, verwendeten wir bereits publizierte Primärdaten einer klinischen Studie.4 Bei der erneuten Analyse mit raspir3 stellte sich heraus, dass bei gesunden Kindern die meisten Bakterien mit hoher und geringer Abundanz ein persistierendes Hintergrundnetzwerk bildeten (Abb.2, links). Die Hintergrundbakterien waren also bereits im ersten Lebensjahr nachweisbar und blieben im Kleinkind- und Vorschulalter vorhanden. Abhängig vom Entwicklungsstadium des Kindes tauchten weitere nichtpersistierende Bakterien in den Atemwegen auf. Bei den CF-Kleinkindern hingegen waren nur wenige persistierende abundante und geringabundante Bakterien im Hintergrund zu detektieren (Abb.2, rechts). Die meisten geringabundanten Bakterien waren dem Kleinkindalter (2–3 Jahre) zuzuordnen. Daraus lässt sich schließen, dass das Immunsystem gesunder Kinder kontinuierlich von einem ausgeprägten, fortbestehenden Netzwerk aus bakteriellen Atemwegskommensalen stimuliert wird, während das Immunsystem bei CF-Kleinkindern größtenteils von den nichtpersistierenden Bakterien und später den CF-Pathogenen trainiert wird.5 Zur Identifikation der Variablen, die ein gesundes von einem CF-Atemwegsmetagenom unterscheiden, verwendeten wir einen Random-Forest-Klassifikationsalgorithmus basierend auf den Bestandteilen des Atemwegsmetagenoms (abundante und geringabundante Bakterien) und wirtsspezifischen Variablen (Alter, Body-Mass-Index, Lung Clearance Index, Pankreasinsuffizienz vs. Pankreassuffizienz). Dabei stellte sich heraus, dass über 70% der Variablen, die für den Entscheidungsprozess („gesund“ oder „CF“) genutzt wurden, mit den geringabundanten Bakterien assoziiert waren.
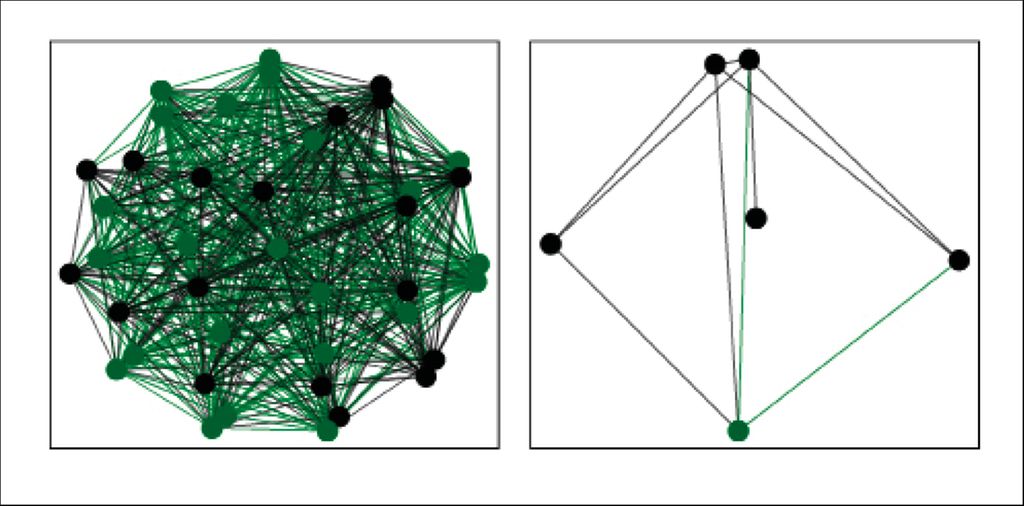
Abb. 2: Das persistierende Hintergrundnetzwerk aus den niedrigabundanten (grün) und abundanten (schwarz) Bakterien bei gesunden (links) und CF-erkrankten (rechts) Kindern
Während abundante Bakterien im Entscheidungsprozess der Klassifizierung eine untergeordnete Rolle spielten, war der Einfluss von wirtsspezifischen Faktoren vernachlässigbar klein. Mithilfe des maschinellen Lernens simulierten wir probiotische Behandlungsansätze, indem wir eine steigende Anzahl von hoch- und geringabundanten Bakterien aus dem gesunden in das unterentwickelte CF-Hintergrundnetzwerk einschleusten und die neu entstandenen Graphstrukturen mit denen des gesunden Netzwerks verglichen. Dabei stellte sich heraus, dass das CF-Bakteriennetzwerk an Stabilität und Widerstandsfähigkeit gewann, wenn gesunde Bakterien mit hoher Speziesdiversität und geringer Dominanz einzelner Spezies in das Netzwerk übertragen wurden. Das originale CF-Netwerk verlor jedoch an Stabilität, wenn einzelne bakterielle Kommensale mit hoher Frequenz in das CF-Netzwerk eingebaut wurden.
Ausblick
Bakterienarten, die in geringer Menge vorkommen, haben eine zentrale Bedeutung im gesunden und erkrankten Atemwegsmikrobiom und sollten daher in zukünftigen Mikrobiomanalysen berücksichtigt werden. Unsere korrelationsbasierten In-silico-Simulationsstudien geben einen ersten Hinweis darauf, dass abhängig von der Art eines probiotischen Behandlungsansatzes das bereits unterentwickelte CF-Hintergrundnetzwerk aus bakteriellen Kommensalen stabilisiert, aber auch weiter destabilisiert werden könnte. Im nächsten Schritt ist es notwendig, mithilfe von komplexen In-vitro- und In-vivo-Modellen die Kausalität unserer Analysen zu überprüfen.
Literatur:
1 Ma ZS: Power law analysis of the human microbiome. Mol Ecol 2015; 24 (21): 5428-45 2 Jousset A et al.: Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J 2017; 11 (4): 853-62 3 Pust MM, Tümmler B: Identification of core and rare species in metagenome samples based on shotgun metagenomic sequencing, Fourier transforms and spectral comparisons. ISME COMMUN 2021; 1: 2 4 Pust MM et al.: The human respiratory tract microbial community structures in healthy and cystic fibrosis infants. NPJ Biofilms Microbiomes 2020; 6 (1): 61 5 Coburn B et al.: Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep 2015; 5: 10241
Das könnte Sie auch interessieren:

Wenig genützte Chance: COPD-Therapie abseits der Medikamente
Neben der medikamentösen Behandlung spielen im Management der COPD nicht-medikamentöse Maßnahmen eine wichtige Rolle. Dies betrifft vor allem die pulmonale Rehabilitation, die ...

Gewebeschädigung: Proteasen bahnen der Allergie den Weg
Warum entwickeln manche Menschen Allergien und andere nicht? Viele Aspekte dieser Frage sind nach wie vor ungeklärt. Auf der klinischen Seite zeigt sich zunehmend, dass die Behandlung ...
