
Der diagnostische Ansatz bei pulmonaler Hypertonie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die pulmonale Hypertonie (PH) ist eine Erkrankung, die oft erst spät erkannt wird: Unspezifische Symptome verschleiern, dass ein erhöhter Druck in der Lungenarterie bereits seit Jahren besteht. Die aktuellen ESC/ERS-Leitlinien definieren klare Diagnosekriterien und betonen die Bedeutung einer präzisen Einordnung in die fünf PH-Gruppen, die entscheidend für Therapie und Prognose sind. Besonders wichtig ist es, in der Diagnostik an die PH zu denken.
Keypoints
-
Symptome von Lungenhochdruck sind ähnlich denen von Asthma, COPD, Dekonditionierung oder psychiatrischen Erkrankungen. Da präkapilläre PH selten ist, wird meistens nicht an die Diagnose gedacht.
-
Übermäßiges Vertrauen in Echokardiografie kann dazu führen, dass PH überschätzt oder unterschätzt wird. Die invasive Bestätigung ist unbedingt erforderlich.
-
Fehlklassifizierungen sind häufig, da PAWP oft nicht korrekt gemessen wird und damit die Unterscheidung zwischen Gruppe 1 (PAH) und Gruppe 2 (Linksherzerkrankung) falsch getroffen wird.
-
Es ist sinnvoll, die Vortestwahrscheinlichkeit für die richtige PH-Diagnose in die Evaluierung miteinfließen zu lassen.
Einleitung
Die pulmonale Hypertonie (PH) ist eine Erkrankung, die durch einen erhöhten Druck in der Lungenarterie gekennzeichnet ist und durch eine Vielzahl von kardiovaskulären, pulmonalen und systemischen Erkrankungen verursacht werden kann. Die ESC/ERS-Leitlinien aus dem Jahr 2022 definieren PH durch die Erhöhung des mittleren Pulmonalarteriendrucks (mPAP) >20mmHg in Ruhe, gemessen mittels Rechtsherzkatheterisierung (RHC), und die präkapillären Formen von Lungenhochdruck mit einem pulmonalen Gefäßwiderstand (PVR) >2 Wood-Einheiten (Prototyp: pulmonal-arterielle Form des Lungenhochdrucks, PAH, Gruppe 1). Weil nur unspezifische Symptome vorliegen, wird PAH praktisch immer spät im Krankheitsverlauf diagnostiziert. Oft besteht PAH seit Jahrzehnten, bevor sie klinisch auffällig wird, zum Beispiel durch eine Schwangerschaft.
Klinischer Verdacht
Der Diagnoseprozess beginnt mit der Identifizierung von Risikopatienten. Symptome, wie zunehmende Belastungsdyspnoe, schwere Beine, Müdigkeit, Brustschmerzen, Herzklopfen, Synkope, treten erst spät auf, wenn schon mehr als die Hälfte der Lungengefäße verloren gegangen sind. Klinisch-physikalisch besteht ein lauter zweiter Herzton, ein rechtsventrikulär hebender Spitzenstoß, ein systolisches Trikuspidalinsuffizienzgeräusch, eine juguläre pulsierende Venenerweiterung, Hepatomegalie, Ödeme, Zyanose. Diese Zeichen findet man allerdings auch erst in fortgeschrittenen Lungenhochdrucksstadien. Risikofaktoren für PAH sind Bindegewebserkrankungen (zum Beispiel systemische Sklerose oder systemischer Lupus erythematodes), angeborene Herzfehler, Leberzirrhose, HIV-Infektion und familiäre Vorbelastung mit pulmonal-arterieller Hypertonie (PAH).
Schrittweiser Diagnosealgorithmus
1. Klinische Untersuchung
Anamnese und körperliche Untersuchung entsprechen einer umfassenden internistischen Abklärung. Grundlegende Laboruntersuchungen beinhalten ein großes Blutbild, Nieren- und Leberfunktion, Schilddrüsenfunktion, BNP/NT-proBNP, metabolische Parameter wie HbA1c und Harnsäure. Im EKG fallen eine Rechtsachsenabweichung, rechtsventrikuläre Hypertrophie, Vergrößerung des rechten Vorhofs, Arrhythmien auf, manchmal ein Rechtsschenkelblock oder hohe R-Zacken in V1/2. In der Röntgenaufnahme des Brustkorbs sieht man vergrößerte Lungenarterien, eine Vergrößerung des rechten Ventrikels und ein verstrichenes aorto-pulmonales Fenster.
2. Echokardiografie
Die transthorakale Echokardiografie (TTE) ist das nichtinvasive Instrument der ersten Wahl. Anhand der Geschwindigkeit des trikuspidalen Regurgitations-Jets wird der systolische Pulmonalarteriendruck geschätzt, und daraus die Wahrscheinlichkeit einer PH in niedrig, mittel, oder hoch stratifiziert. Bei einer trikuspidalen Regurgitationsgeschwindigkeit von >2,9m/sec ist eine hohe echokardiografische Wahrscheinlichkeit für Lungenhochdruck definiert. Dazu müssen auch die Größe/Funktion des rechten Ventrikels, die Abflachung des Septums, die Abmessungen des rechten Vorhofs und der Pulmonalarterie sowie der Hohlvene miteinbezogen werden. Ein Perikarderguss ist prognostisch ungünstig.
3. Lungenfunktionstests (PFTs) und Bildgebung
Spirometrie (Abb. 1) und DLCO (Diffusionskapazität für Kohlenmonoxid) helfen zur Erkennung von COPD, interstitiellen Lungenerkrankungen oder Hypoventilationssyndromen. Hochauflösende CT-Thoraxuntersuchungen sind unerlässlich zur Erkennung von parenchymatösen Lungenerkrankungen, Emphysem und Fibrose. Ventilations-/Perfusions-Scan (V/Q-Scan) ist empfohlen bei allen Patienten mit Verdacht auf PH zum Screening auf CTEPH, da eine hohe Sensitivität für Perfusionsdefekte besteht (Abb. 2). CT-Lungenangiografie (CTPA) liefert anatomische Details bei Verdacht auf CTEPH oder akute Lungenembolie. Die Schwäche der CTPA ist ihre schlechte Auflösung in distalen kleinen Gefäßen.
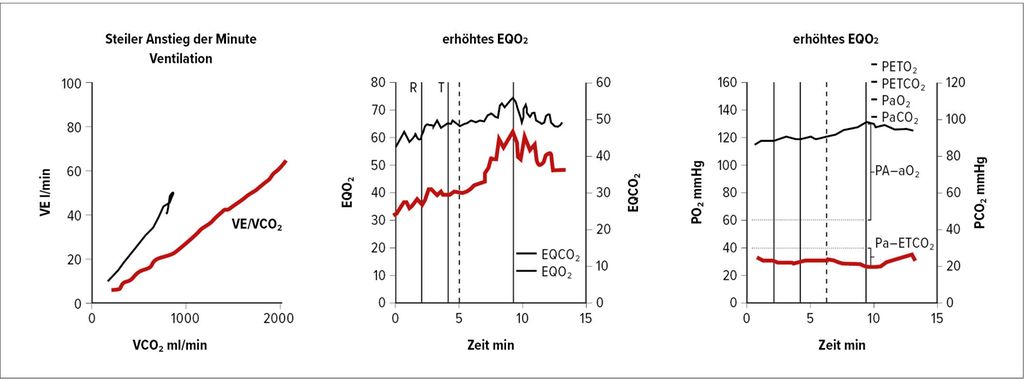
Abb. 1: Von links nach rechts Panels 4, 6 und 9 der Wassermannplots bei Lungengefäßerkrankung. In der Abbildung zeigen rote Kurvenlinien die Lungengefäßerkrankung an, die schwarzen Kurven stammen von normalen Kontrollen. EQO2 steht für ventilatorisches Sauerstoff-Äquivalent berechnet als Atemminutenvolumen/Sauerstoffaufnahme (VO₂), PETCO₂ steht für endtidales CO₂
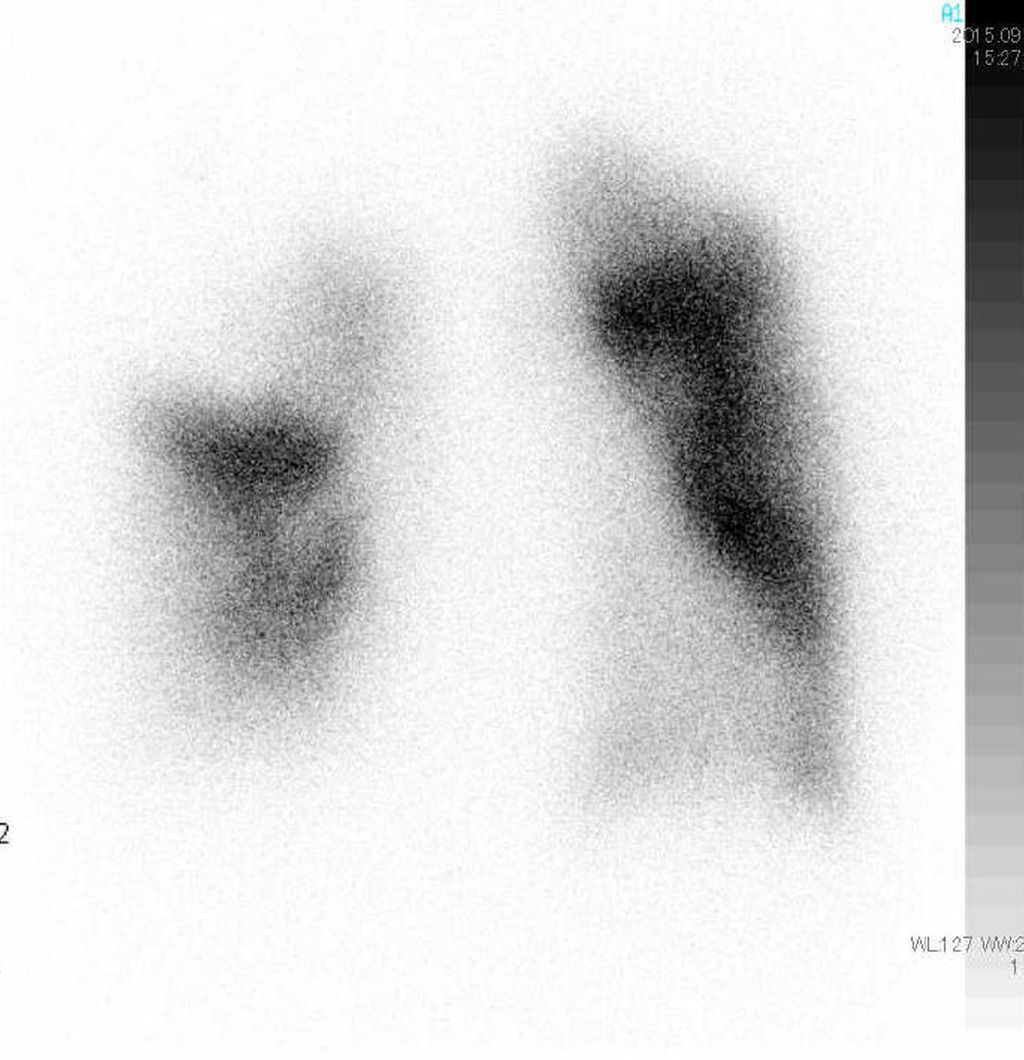
Abb. 2: Anterior-posteriores Projektions-Bild einer Perfusionsszintigraphie bei Chronisch Thromboembolischer Pulmonaler Hypertension (CTEPH). Es zeigen sich typische keilförmige Defekte. Die dazugehörige Ventilationsszintigraphie ist normal, und wird oft übersprungen. Die Perfusionsszintigraphie ist der sensitivste Screeningtest für CTEPH, beweist aber die Erkrankung nicht.
4. Laboruntersuchungen zur Abklärung spezifischer Ursachen
Ein Autoimmunpanel mit ANA, ENA, Antizentromer-Antikörpern, Anti-Scl-70 und der Bestimmung von leichten und schweren Immunglobulin-Ketten im Harn dient der Diagnostik von Bindegewebserkrankungen.
HIV-Test, Hepatitis-Serologie und Leberultraschall helfen bei der Abklärung einer portalen Hypertonie. Gentests sollten bei allen Patienten mit idiopathischer, drogeninduzierter und erblicher PAH durchgeführt werden (z.B. Bestimmung von BMPR2-Mutationen oder biallelischen Mutationen von EIF2KA4). Mutationen im EIF2AK4-Gen, das die GCN2-Kinase codiert, führen zu „pulmonary veno-occlusive disease“ (PVOD) und „pulmonary capillary hemangiomatosis“ (PCH).
5. Kardiopulmonale Belastungsuntersuchung (CPET)
CPET kann zwischen kardialen und pulmonalen Einschränkungen unterscheiden und deckt frühe pulmonale Gefäßerkrankungen auf. CPET erlaubt die Früherkennung von belastungsinduzierter PH bei Patienten mit hohem Krankheitsrisiko, z.B. homozygote Träger von BMPR2-Mutationen, ist aber wenig spezifisch.
6. Rechtsherzkatheteruntersuchung
Die Rechtsherzkatheterisierung (RHC) ist obligatorisch zur Bestätigung einer Lungenhochdrucks-Diagnose, zur Definition des hämodynamischen Typs, zur hämodynamischen Austestung und später zur Steuerung der Therapie. Es werden die systolischen, diastolischen und mittleren Drucke im rechten Vorhof, Ventrikel, in der Pulmonalarterie und in der Wedge Position als pulmonaler arteriolärer Verschlussdruck (PAWP) gemessen, das Herzzeitvolumen, die respektiven Sauerstoffsättigungen und der Lungengefäßwiderstand (PVR) berechnet. Der pulmonale Vasoreaktivitätstest mit inhalativem Stickoxid ist indiziert bei idiopathischer, erblicher und medikamentös induzierter PAH, um Kandidaten für eine orale Kalziumkanalblocker-Therapie zu identifizieren.
Aus den Daten des Rechtsherzkatheters wird klassifiziert:
-
Präkapilläre PH: mPAP >20mmHg, PCWP ≤15mmHg, PVR >2 WU
-
Postkapilläre PH: mPAP >20mmHg, PCWP >15mmHg.
Differenzialdiagnose und Klassifizierung
Der Diagnoseprozess gipfelt in der Zuordnung der Enddiagnose zu einer der fünf PH-Gruppen:
-
Gruppe 1 – pulmonale arterielle Hypertonie (PAH): idiopathisch, erblich, medikamentös induziert, assoziiert mit Bindegewebserkrankung, HIV, kongenitaler Herzerkrankung, portale Hypertonie.
-
Gruppe 2 – PH assoziiert mit Linksherzerkrankung: LV-systolische/diastolische Dysfunktion, Herzklappenerkrankung.
-
Gruppe 3 – PH assoziiert mit Lungenerkrankungen/Hypoxie: COPD, interstitielle Lungenerkrankung, OSA, alveoläre Hypoventilation.
-
Gruppe 4 – PH assoziiert mit Pulmonalarterienobstruktionen: CTEPH, andere vaskuläre Obstruktionen.
-
Gruppe 5 – PH mit unklaren oder multifaktoriellen Mechanismen: hämatologische, metabolische, systemische Störungen.
Fallstricke und Herausforderungen
Symptome von Lungenhochdruck sind ähnlich denen von Asthma, COPD, Dekonditionierung oder psychiatrischen Erkrankungen. Da präkapilläre PH selten ist (Inzident 1–5 pro Million und Jahr), wird meistens nicht an die Diagnose gedacht. Andererseits führt ein übermäßiges Vertrauen in Echokardiografie dazu, dass PH überschätzt oder unterschätzt wird. Eine invasive Bestätigung ist unbedingt erforderlich. Fehlklassifizierungen sind häufig, da PAWP oft nicht korrekt gemessen wird, und damit die Unterscheidung zwischen Gruppe 1 (PAH) und Gruppe 2 (Linksherzerkrankung) falsch getroffen wird. Es ist sinnvoll, die Vortestwahrscheinlichkeit für die richtige PH-Diagnose in die Evaluierung miteinfließen zu lassen.
Fazit
Der diagnostische Ansatz bei pulmonaler Hypertonie ist systematisch und folgt dem klinischen Verdacht mit einem strukturierten Algorithmus nichtinvasiver Tests, gefolgt von einer bestätigenden Rechtsherzkatheteruntersuchung. Die korrekte Einstufung in eine der fünf PH-Gruppen ist wichtig, da sie direkten Einfluss auf die Therapie und Prognose hat. Eine multidisziplinäre Beurteilung in spezialisierten PH-Zentren wird dringend empfohlen, insbesondere bei Patienten mit Verdacht auf PAH oder CTEPH. THINK PH!
Literatur:
bei der Verfasserin
Potenzielle Interessenkonflikte:
Irene Lang erhielt Forschungsgrants von AOP-Health und Sprecher- sowie Beraterhonorare von AOP-Health, Janssen, MSD, United Therapeutics, Boehringer Ingelheim, Amarin und Novo-Nordisk.
Das könnte Sie auch interessieren:
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

Studienergebnisse mit Praxisrelevanz
Die diesjährigen Scientific Sessions der US-Kardiologengesellschaft American College of Cardiology (ACC.26) fanden von 28. bis 30. März in New Orleans statt. In insgesamt sieben Late- ...

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...