
Medikamentöse Therapieoptionen bei Akromegalie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Wir beschreiben in dieser Kasuistik den langen Weg einer jungen Patientin mit Akromegalie bis zur erfolgreichen Therapie. Darüber hinaus möchten wir die Symptomatik, Diagnostik und Therapieoptionen vermitteln. Wir erklären, wie Therapieziele erreicht werden und damit die Lebensqualität der Patientinnen und Patienten entscheidend verbessert werden können.
Keypoints
-
Für eine Akromegalie verantwortlich ist die Überproduktion des Wachstumshormons.
-
Häufigste Ursache ist ein Hypophysentumor (Adenom). Ab einer Grösse von 10 mm spricht man von einem Makroadenom. Eine ektope Wachstumshormonproduktion ist eine Rarität.
-
Nach der Labordiagnostik erfolgt die radiologische Abklärung mittels MRT der Hypophysen-/Sellaregion.
-
Die einzige kurative Therapie ist die operative Sanierung.
-
Medikamentöse Therapieoptionen sind Dopaminagonisten, Somatostatin-Analoga und Wachstumshormonrezeptor- Antagonisten.
Bei unserer Patientin wurde in ihrem 25. Lebensjahr, im Jahr 2008, eine Akromegalie diagnostiziert. In der Anamnese aufgefallen waren vor allem ein starker Kopfschmerz, Zyklusstörungen sowie Schmerzen in den Fingern (Tab. 1).

Tab. 1: Häufige Symptome bei Akromegalie
Laborchemisch bestand eine Erhöhung des Wachstumshormons (STH: 98µIU/ml; Norm: 0,2–10,0µIU/ml), des Insulin-like Growth Factor 1 (IGF-1: 1362ng/ml; altersentsprechende Norm: 113–450ng/ml) und des Prolaktins (770µIU/ml; N: 113–450µIU/ml). In der kranialen Magnetresonanztomografie zeigte sich ein Makroadenom der Hypophyse (2,7x1,6x1,5cm).
Eine operative Sanierung erfolgte im Februar 2009, postoperativ zeigten sich jedoch weiterhin stark erhöhte STH- und IGF-1-Werte. Eine optimale medikamentöse Kontrolle (IGF-1 im Normbereich) konnte erst Jahre später nach Ausschöpfung aller Optionen erreicht werden.
Akromegalie: Symptome und Diagnostik
Die für die Akromegalie verantwortliche Überproduktion des Wachstumshormons (somatotropes Hormon, STH) stimuliert in weiterer Folge die Bildung von IGF-1 in der Leber, welches das Zellwachstum fördert und so zu einer Vielzahl von morphologischen und metabolischen Veränderungen führt, die sich teilweise sehr diskret entwickeln. Der klinische Zusammenhang ergibt sich meist erst retrospektiv, weshalb häufig Jahre vergehen zwischen Erkrankungsbeginn und Diagnosestellung. Peripher kann es durch die Stimulierung des Zellwachstums zu Gelenksbeschwerden aufgrund einer hypertrophen Arthropathie sowie zu einem Karpaltunnelsyndrom kommen. Häufig wird auch von vermehrtem Schwitzen berichtet sowie von geschwollenen Füssen und Fingern (Ringe passen nicht mehr).
Als Komorbiditäten können sich eine arterielle Hypertonie, eine Kardiomyopathie, ein Schlafapnoe-Syndrom sowie eine gestörte Glukosetoleranz bis hin zum Diabetes mellitus entwickeln. Die typischen äusserlichen Veränderung mit Vergrösserung der Mandibula mit Prognathie, Verbreiterung der Zahnzwischenräume sowie Verbreiterung der Nasenwurzel und prominenter Supraorbitalwülste werden von Patientinnen und Patienten sowie deren Angehörigen aufgrund der langsamen Progression häufig vorerst nicht wahrgenommen und zeigen sich dann im Vergleich mit älteren Fotos (z.B. Passbild). Es sind aber gerade diese äusserlichen Veränderungen, die zu der zum Teil stark eingeschränkten Lebensqualität von Akromegaliepatienten beitragen können. Tritt eine vermehrte Wachstumshormonproduktion im Kindesalter vor dem knöchernen Schluss der Epiphysenfugen auf, kommt es zum klinischen Bild des Gigantismus.
Ein Hypophysentumor, meist ein Adenom, ist die häufigste Ursache einer Akromegalie; eine ektope Wachstumshormonproduktion ist eine Rarität. Bei einem Adenom über 10mm spricht man von einem Makroadenom.
Symptome, verursacht durch das lokale Wachstum, können ebenfalls zur Diagnose führen, wie dies bei unserer Patientin der Fall war. Im Vordergrund stehen dabei Kopfschmerzen sowie Gesichtsfeldausfälle (bitemporale Hemianopsie u.a.), sofern das Chiasma opticum komprimiert wird. Zusätzlich kann es zum Ausfall (Hypogonadismus, sekundäre Hypothyreose, sekundäre Nebennierenrindeninsuffizienz) oder zur Überproduktion (reaktive Hyperprolaktinämie) weiterer hypophysärer Hormone mit entsprechender Symptomatik kommen.
Ein IGF-1-Wert im altersabhängigen Normbereich schliesst eine Akromegalie weitgehend aus, das STH ist aufgrund seiner pulsatilen Ausschüttung und kurzen Halbwertszeit als Screeningtest nicht geeignet. Zusätzlich soll zur Diagnosesicherung ein oraler Glukosetoleranztest (oGTT) durchgeführt werden. Physiologischerweise supprimiert ein Blutzuckeranstieg die Ausschüttung von STH. Bei einer autonomen Sekretion zeigen sich zwei Stunden nach Glukosegabe eine fehlende Supprimierbarkeit oder sogar ein paradoxer STH-Anstieg.
Nach der Labordiagnostik erfolgt die radiologische Abklärung, in der Regel mit der MRT der Hypophysen-/Sellaregion.
Therapeutisch steht die operative Sanierung an erster Stelle und sie ist auch der einzige kurative Ansatz. Ziele sind eine biochemische Heilung der Akromegalie mit postoperativen IGF-1-Spiegeln im Normbereich und supprimierten STH-Werten im oGTT sowie eine Besserung der klinischen Symptomatik und des radiologischen Befundes. Mit dem Erreichen dieser Zielwerte ist auch eine Normalisierung der Lebenserwartung verbunden.
Falls eine kurative Operation nicht möglich ist (bei Makroadenomen in ca. 50% der Fälle), stehen folgende medikamentöse Therapieoptionen zur Verfügung: Dopaminagonisten, Somatostatin-Analoga und Wachstumshormonrezeptor-Antagonisten (Abb. 1).
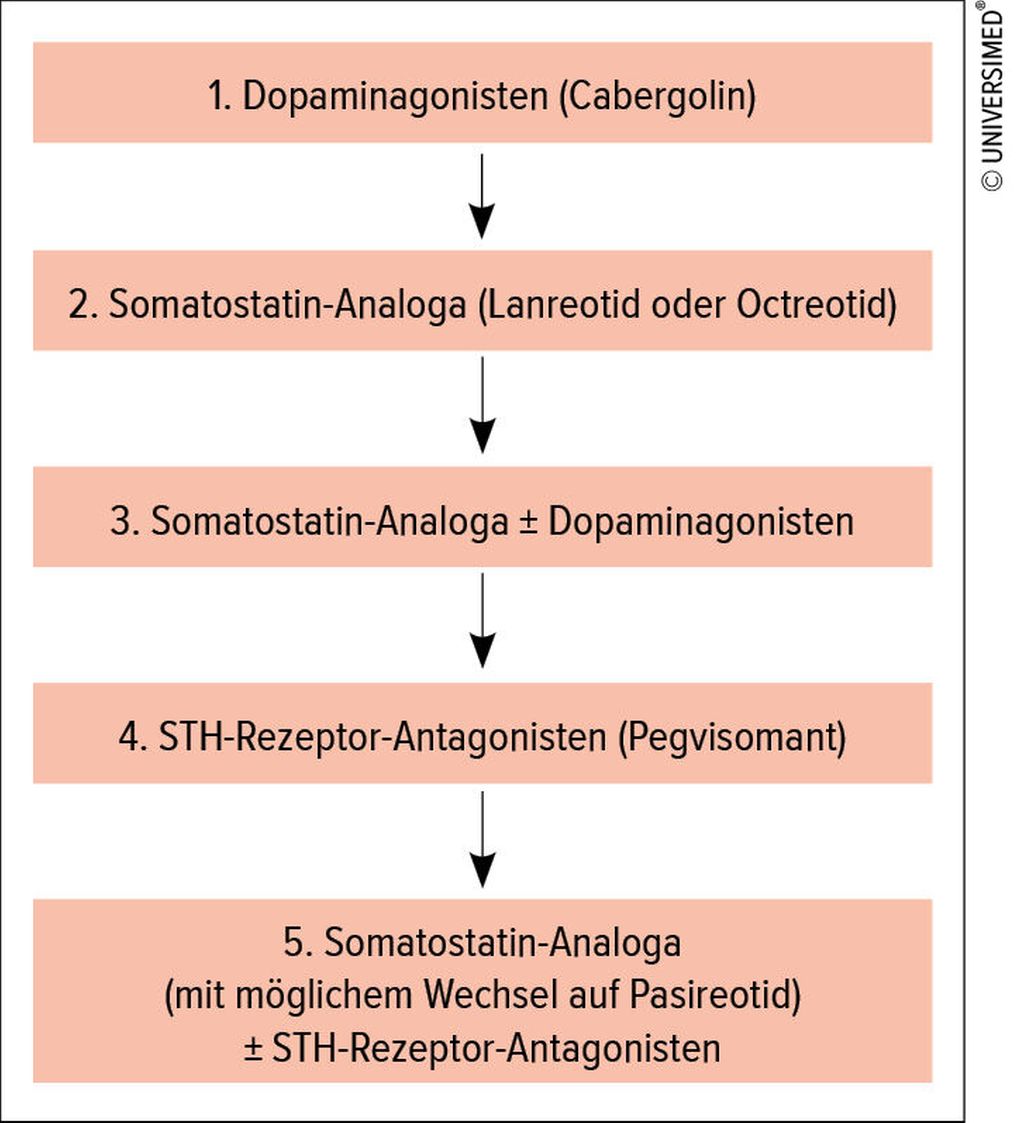
Abb. 1: Medikamentöse Optionen bei Akromegalie (vereinfacht)
Verlauf und Therapie
Bei unserer Patientin bestanden drei Monate nach erfolgter Operation weiterhin stark erhöhte IGF-1-Werte (629ng/ml), zusätzlich wurde in den radiologischen Kontrollen eine 5mm grosse postoperative Struktur beschrieben, am ehesten Granulationsgewebe entsprechend (DD: Resttumor oder Rezidiv) .
Dopaminagonisten bilden einen ersten medikamentösen Schritt und auch die einzige orale Therapieoption. Vor allem bei Adenomen mit zusätzlicher Prolaktinsekretion und postoperativ nur mässig ausgeprägter Restaktivität (IGF-1 weniger als das Doppelte des Normbereichs) kann sich ein Versuch mit Dopaminagonisten lohnen, wobei sich in den Studien Cabergolin (Dostinex®) potenter als Bromocriptin zeigte. Häufige Nebenwirkungen sind Müdigkeit, Kopfschmerzen, gastrointestinale Beschwerden, Orthostase sowie depressive Verstimmung.
Bei unserer Patientin wurde mit Cabergolin zweimal pro Woche begonnen, worunter der IGF-1-Spiegel vorerst sank, jedoch auch nach einem Jahr Therapie nicht den altersabhängigen Normbereich erreichte. Zusätzlich war das STH im oGTT nicht supprimierbar, sodass als nächster Schritt ein Somatostatin-Analogon dazugegeben wurde.
Die Ausschüttung von STH wird durch Somatostatin inhibiert, wobei es fünf bekannte Rezeptoruntertypen für Somatostatin gibt. Synthetische Somatostatin-Analoga mit längerer Halbwertszeit sind Octreotid (Sandostatin® LAR) und Lanreotid (Somatuline Autogel®), welche vor allem an den Somatostatin-Rezeptorsubtypen 2 und 5 binden. Vor allem Depotpräparate, welche einmal im Monat tief subkutan (Lanreotid) bzw. intramuskulär (Octreotid) gegeben werden, sind eine dauerhafte Therapie bei Akromegalie. Für diese Anwendung ist auch eine tumorverkleinernde Wirkung beschrieben. Bei ausgeprägter Symptomatik und stark erhöhten IGF-1-Werten kann initial bereits mit dieser Therapie begonnen werden. Ausserdem ist eine Kombination von Dopaminagonisten mit Somatostatin-Analoga möglich.
Unsere Patientin erhielt in weiterer Folge Lanreotid zusätzlich zu Cabergolin. Die Dosis konnte bei guter Verträglichkeit von anfangs 60mg auf 120mg im Monat gesteigert werden. Als typische Beschwerden, die jedoch zum grössten Teil vorübergehend sind, können gastrointestinale Symptome wie Blähungen, Übelkeit, Diarrhö oder Erbrechen auftreten. Zusätzlich ist eine Verschlechterung des Glukosestoffwechsels möglich und ein erhöhtes Risiko für Gallensteine ist bekannt. Doch auch unter dieser Therapie zeigten sich die IGF-1-Werte unserer Patientin konstant erhöht mit sogar leicht steigender Tendenz.
Als nächste Option steht die Gabe eines Wachstumshormonrezeptor-Antagonisten – Pegvisomant (Somavert®), das täglich subkutan verabreicht werden muss – zur Verfügung. Dieses bindet an den Wachstumshormonrezeptor, ohne dass eine Signaltransduktion ausgelöst wird, sodass die Bildung von IGF-1 ausbleibt.
Bei unserer Patientin wurde im Mai 2011 mit Pegvisomant begonnen, Lanreotid wurde pausiert. Mit dieser Therapie konnte zum ersten Mal eine Reduktion von IGF-1 in den Normbereich erzielt werden. Zu einem Anstieg der Transaminasen, was unter Pegvisomant-Therapie möglich ist und in seltenen Fällen dazu führt, dass die Medikation abgesetzt werden muss, kam es bei unserer Patientin nicht. Da Pegvisomant zwar häufig IGF-1 ausreichend gut supprimiert, aber keinen tumorverkleinernden Effekt aufweist, sind regelmässige radiologische Kontrollen besonders wichtig. Bei unserer Patientin zeigte sich in den radiologischen Verlaufskontrollen kein Wachstum des fraglichen Resttumors, die IGF-1-Werte blieben unter 10mg Pegvisomant stabil.
Kinderwunsch bei Akromegalie
Zu diesem Zeitpunkt bestand vonseiten der Patientin ein Kinderwunsch. Bei einer gut kontrollierten Akromegalie ist eine Schwangerschaft möglich, die medikamentöse Therapie muss jedoch dafür pausiert werden. Eine Gabe von kurz wirksamem Octreotid ist nur in Ausnahmefällen möglich.
Im Jahr 2013 wurde unsere Patientin schwanger und die bestehende Therapie pausiert. In den Kontrollen zeigten sich steigende IGF-1-Werte, radiologisch zeigte sich das bekannte Restgewebe ohne Dynamik. Die Patientin selbst gab während der Schwangerschaft starke Kopfschmerzen und Schmerzen in den Händen an, welche gegen Ende der Schwangerschaft kaum auszuhalten waren. Bis auf einen Gestationsdiabetes verlief die Schwangerschaft jedoch komplikationslos und die Patientin brachte 2014/02 einen gesunden Sohn zur Welt.
Weiterer Verlauf
Nach der Entbindung wurde 2014 erneut mit Pegvisomant begonnen. Da in den Kontrollen bis 2016 trotz Dosissteigerung nur einmal ein IGF-1-Wert im Normbereich festgestellt werden konnte, entschieden wir uns dafür, der Patientin erneut Lanreotid zusätzlich zur bestehenden Therapie zu gegeben. Aufgrund der unterschiedlichen Wirkmechanismen können Somatostatin-Analoga und Pegvisomant als Wachstumshormonrezeptor-Antagonist kombiniert werden. Im weiteren Verlauf erfolgten eine maximale Aufdosierung dieser medikamentösen Therapie und die zusätzliche Gabe von Cabergolin. Die Patientin verabreichte sich selbst s.c. sowohl Pegvisomant als auch Lanreotid (alle 4 Wochen) und führte die Therapie sehr exakt durch, trotzdem liess sich keine Suppression von IGF-1 in den Normbereich erzielen.
Mit Pasireotid (Signifor®) steht seit einigen Jahren ein weiteres Somatostatin-Analogon zur Verfügung, welches im Gegensatz zu Octreotid und Lanreotid auch an den Somatostatin-Rezeptorsubtypen 1 und 3 sowie stärker am Subtyp 5 wirkt. In Studien konnten bei Patienten mit nicht ausreichend kontrollierter Akromegalie nach Gabe von Lanreotid oder Octreotid durch einen Wechsel auf Pasireotid sowohl der IGF-1-Wert als auch der STH-Spiegel gesenkt werden. Bei Pasireotid ist das Risiko für die Entwicklung eines Diabetes mellitus höher als unter Lanreotid oder Octreotid, sodass eine entsprechende Aufklärung der Patientinnen und Patienten sowie regelmässige Kontrollen des Glukosestoffwechsels wichtig sind. Weiterhin ist auf eine mögliche Verlängerung der QTc-Zeit zu achten sowie auf die Induktion von Gallensteinen.
Im August 2018 erfolgte bei unserer Patientin ein Wechsel von Lanreotid auf Pasireotid. Bei der ersten Kontrolle im November 2018 zeigte sich bereits ein Rückgang von IGF-1 auf 216ng/ml in den Normbereich (81–278ng/ml) gegenüber 378ng/ml, die im März 2018 noch vor der Gabe von Pasireotid festgestellt wurden. Im Februar 2019 betrug die IGF-1-Konzentration 153ng/ml, was einer biochemischen Heilung entspricht. Zusätzlich gab die Patientin an, dass ihre Kopfschmerzen seit dem Beginn der Therapie deutlich besser geworden sind, was sie als sehr grossen Zugewinn an Lebensqualität sieht. Zur Erfassung der Lebensqualität bei Akromegaliepatienten stehen Fragebögen zur Option (z.B. AcroQol).
In der MRT vom Mai 2018 hatte sich am Sellaboden eine gegenüber den Vorbefunden gering an Grösse zugenommene Struktur (fragl. Resttumor 6mm) gezeigt, welche im Juli 2019 (MRT am selben Institut) nicht mehr nachweisbar war. Eine anfängliche Diarrhö als bekannte Nebenwirkung besserte sich im Verlauf. Mit dem Erreichen der Therapieziele zeigte unsere Patientin ein knappes Jahr nach Therapiebeginn mit Pasireotid einen hoffnungsvollen Verlauf, was sich auch in ihrer Lebensqualität widerspiegelte.
Autoren:
Dr. med. Greta Gericke
Univ.-Doz. Dr. med. Christoph Schnack
1. Medizinische Abteilung
Krankenanstalt Rudolfstiftung, Wien
E-Mail: christoph.schnack@wienkav.at
Literatur:
bei den Verfassern
Das könnte Sie auch interessieren:

Diabetes erhöht das Sturzrisiko deutlich
Eine dänische Studie kommt zu dem Ergebnis, dass sowohl Patienten mit Typ-1- als auch Patienten mit Typ-2-Diabetes öfter stürzen und häufiger Frakturen erleiden als Menschen aus einer ...

Neue Studiendaten zu Typ-2-Diabetes und Lebensstil
Dass gesunde Ernährung und Bewegung das Diabetesrisiko sowie verschiedene Risiken von Patienten mit Diabetes senken, ist seit Langem bekannt. Und das Detailwissen zur Bedeutung von ...

Wie oft wird Diabetes nicht oder spät erkannt?
Im Allgemeinen wird von einer hohen Dunkelziffer an Personen mit undiagnostiziertem Typ-2-Diabetes ausgegangen. Ein Teil davon sind von Ärzten „übersehene“ Fälle. Eine von der University ...