Gentherapeutische Ansätze für die Epidermolysis bullosa
Die Anwendung einer Gentherapie ist die einzige Möglichkeit, die genetische Ursache einer Erkrankung zu bekämpfen. Bis dato fand alleine die Genersatztherapie als therapeutischer Ansatz für Genodermatosen ihren Weg in die Klinik. Steht „Gene editing“ für die Behandlung von genetischen Hauterkrankungen ante portas?
Keypoints
-
Genodermatosen sind genetisch bedingte Hauterkrankungen, die durch Genmutationen verursacht werden.
-
Epidermolysis bullosa ist eine Genodermatose, die autosomal-dominant oder -rezessiv vererbt wird.
-
Bei der Ex-vivo-Gentherapie werden Patientenzellen außerhalb des Körpers korrigiert.
-
Bei der Ex-vivo-Genersatztherapie wird das mutierte Gen durch eine in die Zelle eingebrachte gesunde Genkopie ergänzt.
-
Über Ex-vivo-„Gene editing“ mittels Designer-Nukleasen kann eine punktuelle Genkorrektur im Erbgut erreicht werden.
Die ersten Schritte im Bereich der Gentherapie für angeborene genetische Erkrankungen wurden in den 1990er-Jahren getätigt und von der Öffentlichkeit gespannt verfolgt. Die frühe Euphorie erhielt aber im Zuge einer gentherapeutischen Behandlung der Immunschwächeerkrankung X-SCID („severe combined immunodeficiency“) einen Dämpfer. Mehrere Kinder entwickelten aufgrund der Behandlung eine Leukämie. Grund dafür war die Transaktivierung eines Protoonkogens als Folge der viralen Behandlung der Zellen.1 Virale Integrationen in das Genom erfolgen bei der Genersatztherapie eher zufällig und können dadurch Gene im Bereich der Integration zerstören oder deren Expression beeinflussen.
Trotzdem stellt die Haut ein vielversprechendes Zielorgan für eine Gentherapie dar. Mögliche unerwünschte Nebenwirkungen der Behandlung können schnell erkannt und behandelt werden. Unter anderen wurde aufgrund dieser Tatsache eine auf Retroviren basierende Ex-vivo-Gentherapie für die blasenbildende Hauterkrankung Epidermolysis bullosa (EB)2 initiiert und erfolgreich bei drei Patienten mit der junktionalen Form der EB angewandt.3–5 Bis dato sind die behandelten Stellen blasenfrei,6 wie Dr. Michele De Luca berichtet. Der langfristige Erfolg der Applikation wird durch die Korrektur von epidermalen Stammzellen erreicht. Deren Anzahl nimmt im Alter und durch chronische Wunden, wie sie bei der EB vorliegen, ab, wodurch es mit zunehmenden Alter immer schwieriger wird, genug epidermale Stammzellen für eine autologe Stammzelltherapie zu gewinnen.7
Ex-vivo-Gentherapie: vielversprechend, aber verbesserungswürdig
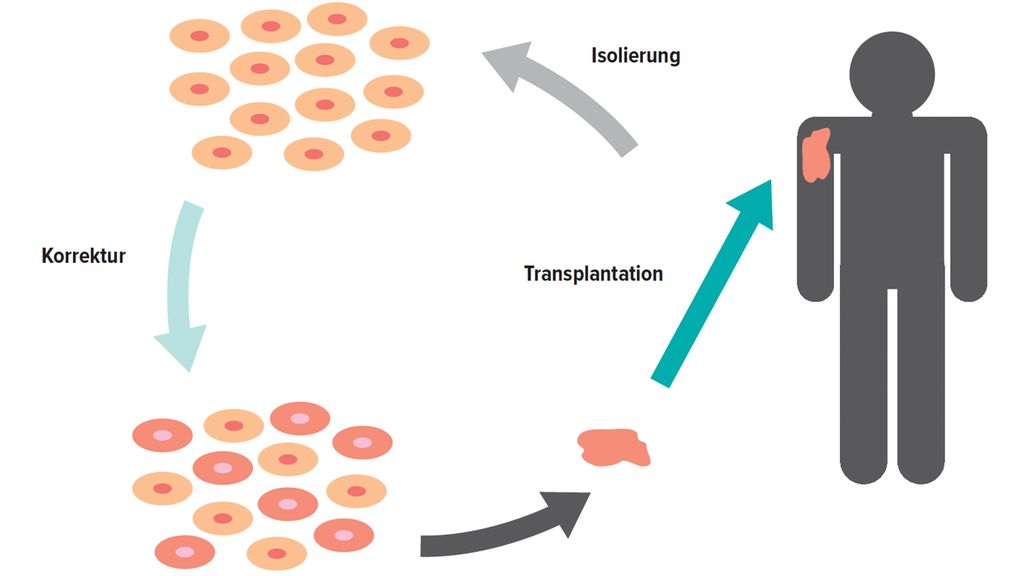
Im Gegensatz zu einer In-vivo-Behandlung findet bei einer Ex-vivo-Gentherapie die Genkorrektur außerhalb des Körpers statt. Nach einer klinischen und genetischen Analyse des Patienten werden Hautstammzellen aus einer vom Patienten bereitgestellten Hautbiopsie isoliert und im Labor weitergezüchtet. Nach der Transfektion der Stammzellen und einer folgenden Sicherheitsanalyse werden die genetisch korrigierten Zellen zu Hautschichten expandiert und dem Patienten rücktransplantiert (Abb. 1).8 Gerade für die monogenetische Hauterkrankung EB stellt die Ex-vivo-Gentherapie eine effiziente Therapieform dar, um die Ursache der Erkrankung lokal zu bekämpfen.
Abb. 1: Durchführung einer Ex-vivo-Gentherapie (modifiziert nach March OP et al. 201912)
Bei der EB führen genetische Veränderungen, auch Mutationen genannt, in Genen, aus denen Strukturproteine der Haut gebildet werden, zu deren Funktionsverlust. Lokalisiert im Bereich der Basalmembranzone (BMZ) der Haut, haben sie eine wichtige stabilisierende Funktion bei der Verknüpfung der Oberhaut (Epidermis) mit der darunterliegenden Lederhaut (Dermis). Die beim Patienten vorliegenden Mutationen in den entsprechenden Genen führen zur funktionellen Beeinträchtigung oder zur vollständigen Abwesenheit des resultierenden Strukturproteins in der Haut. Die folglich destabilisierende Wirkung auf die Hautintegrität geht mit Blasenbildungen und Erosionen an der Haut sowie Schmerz und Juckreiz einher.2
Die bis dato einzigen klinischen Anwendungen bei EB mit Langzeitwirkung basieren auf einer Genersatztherapie.3–5 Um die Funktion des mutierten Gens wiederherzustellen, wird dieses durch eine zusätzlich in die Zelle eingebrachte gesunde Kopie ergänzt. 2015 wurde ein damals siebenjähriger Junge mit der junktionalen EB-Form an der Universitätsklinik Bergmannsheil in Bochum mit einer Ex-vivo-Genersatztherapie erfolgreich behandelt. Der Junge verlor aufgrund einer schweren bakteriellen Infektion über 60% seiner Haut. Nur eine schnell initiierte gentherapeutische Applikation konnte ihm das Leben retten. Die Möglichkeit, über 80% der Haut über dieses Verfahren zu rekonstruieren, untermauerte das große Potenzial einer Ex-vivo-Gentherapie für die Behandlung von Patienten mit monogenetischen Genodermatosen.5 Analog zur Behandlung von Patienten mit der junktionalen Form der EB wurde ein ähnlicher Ansatz für die dystrophe EB-Form gewählt.9 Auch hier wurden Hautstammzellen aus einer Hautbiopsie gewonnen und im Labor kultiviert. Die Patientenzellen wurden dann mit einem retroviralen Vektor, der eine komplette „Collagen type VII alpha 1 chain“(COL7A1)-Genkopie trägt, korrigiert, zu Hautschichten expandiert und dann auf den Patienten transplantiert. Obwohl eine Wiederherstellung der durch Kollagen VII aufgebauten Ankerfibrillen in den Hautschichten ersichtlich war,10 zeigte eine folgende klinische Phase-I/II-Studie eine progressive Verringerung der Kollagen-VII-Produktion innerhalb des ersten Jahres nach der Transplantation.9 Diese Ergebnisse unterstreichen, dass der Erfolg einer Genersatztherapie vom zu ergänzenden Gen/Protein bzw. von der jeweiligen EB-Form abhängig ist.
Noch in den Kinderschuhen: „Gene editing“
Die Entwicklung alternativer Therapieansätze ist aufgrund oben beschriebener Schwierigkeiten dringend notwendig. Das „Gene editing“ basiert aktuell auf über Designer-Nukleasen vermittelten Genkorrekturen, die in der Regel mutationsspezifisch und folglich patientenspezifisch erfolgen. Mit der Entwicklung genspezifischer Zinkfingernukleasen begann das Zeitalter der Designer-Nukleasen, deren Spezifität und Effizienz mit dem Einsatz von „transcription activator-like effector nucleases“ (TALEN) und „clustered regularly interspaced short palindromic repeats“(CRISPR)/„CRISPR associated protein 9“(Cas9)-Nukleasen weiter verbessert werden konnten.11–15 Die Reparatur der Erbschäden über „Gene editing“ wird über das Andocken und Schneiden der DNA durch die entsprechende Nuklease initiiert, wodurch Reparaturmechanismen in der Zelle aktiviert werden. Bei der Erkrankung EB wurde „Gene editing“ für die Inaktivierung von Genen,in denen dominant negative Mutationen für den Phänotyp am Patienten verantwortlich sind,11, 15 oder für die potenziell spurenlose Korrektur über homologe Rekombination13, 15–17 erfolgreich angewandt. Da bekannt ist, dass Designer-Nukleasen an andere Stellen im Genom bzw. Erbgut binden und dort schneiden können,18 bedarf es einer gründlichen Sicherheitsanalyse, bevor eine klinische Anwendung infrage kommt. Zudem wird die Entwicklung von potenziell sichereren Nukleasen13, 16, 19 forciert.
Fazit
Die Genersatztherapie untermauerte als Vorreiter der Ex-vivo-Gentherapie die Möglichkeit, EB-Patienten mit dieser Therapieform zu behandeln. Speziell für die junktionale EB konnten in unabhängigen Studien eine hohe Effizienz und Sicherheit der Anwendung gezeigt werden. Besonders beim ersten Patienten3 konnten in einem langjährigen Follow-up6 keine schweren Nebenwirkungen festgestellt werden.
Da der Therapieerfolg maßgeblich vom zu korrigierenden Gen bzw. von der Art der Vererbung (rezessiv oder dominant) der krankheits-assoziierten Mutation abhängt, ist die Suche nach einer gentherapeutischen Alternative unumgänglich. Designer-Nukleasen, wie die CRISPR/Cas9-Nuklease, können dieses Gentherapiewerkzeug der Zukunft darstellen. Genetische Veränderungen jeglicher Art können genunabhängig punktuell korrigiert werden. Trotz der vielversprechenden Verbesserungen im „Gene editing“-Bereich im Hinblick auf Effizienz und Sicherheit müssen erste klinische Studien zu anderen schweren Erkrankungen (siehe https://clinicaltrials.gov/) die Anwendbarkeit für Genodermatosen überprüfen.
Literatur:
1 Hacein-Bey-Abina S et al.: N Engl J Med 2003; 348(3): 255-6 2 Fine JD et al.: J Am Acad Dermatol 2014; 70(6): 1103-26 3 Mavilio F et al.: Nat Med 2006; 12(12): 1397-402 4 Bauer JW et al.: J Invest Dermatol 2017; 137(3): 778-81 5 Hirsch T et al.: Nature 2017; 551(7680): 327-32 6 De Rosa L et al.: Stem Cell Reports 2013; 2(1): 1-8 7 De Rosa L et al.: Expert Opin Orphan Drugs 2018; 6(4): 283-93 8 Murauer et al.: Keio J Med 2015; 64(2): 21-5 9 Siprashvili Z et al.: JAMA 2017; 316(17): 1808-17 10 Siprashvili Z et al.: Hum Gene Ther 2010; 21(10): 1299-1310 11 Aushev M et al.: Mol Ther Methods Clin 2017; 6: 112-23 12 March OP et al.: J Invest Dermatol 2019; 139(8): 1699-710 13 Kocher T et al.: Mol Ther Nucleic Acids 2019; 18: 496-507 14 Porteus MH: N Engl J Med 2019; 380(10): 947-59 15 March OP et al.: Cells 2020; 9(1): pii: E112 16 Kocher T et al.: Mol Ther 2017; 25(11): 2585-98 17 Hainzl S et al.: Mol Ther 2017; 25(11): 2573-84 18 Fu Y et al.: Nat Biotechnol 2013; 31(9): 822-6 19 Ran FA et al.: Cell 2013; 154(6): 1380-9
Das könnte Sie auch interessieren:
Narbenbehandlung ohne Devices
Für die Behandlung von Narben hat sich der Einsatz moderner Lasertechnologien als effektiv erwiesen. Doch wie kann man den Betroffenen helfen, wenn kein ablativer fraktionierter CO2- ...

Update atopisches Ekzem
In den vergangenen Jahren haben sich das Verständnis des atopischen Ekzems (AE) sowie die therapeutischen Möglichkeiten deutlich weiterentwickelt. Und der Weg ist noch nicht zu Ende ...

Coaching für Ärzt:innen: Klarheit finden, an Stärke gewinnenEinblicke in den Coachingprozess
Die Anforderungen an Ärztinnen und Ärzte in Kliniken und Praxen sind enorm. Neben fachlichem Können werden täglich auch persönliche Ressourcen wie Resilienz und Entscheidungsstärke ...