
Wege zur korrekten Diagnose
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Patientinnen und Patienten mit verhärteter Haut können an sehr unterschiedlichen Erkrankungen leiden. Im Zentrum steht immer ein Fibroblast, eine mesenchymale Vorläuferzelle, die durch interne oder externe Mechanismen angestossen wird und dann entweder exzessiv Kollagen, Muzin oder Paraproteine produziert oder zu proliferieren beginnt. Trotz phänotypischer Ähnlichkeiten lassen sich die daraus resultierenden Hauterkrankungen unterscheiden.
Keypoints
-
Fibrosierende Hauterkrankungen sind hoch heterogen, ihre Prognose variiert stark und hängt vom Umfang der systemischen Manifestation ab. Eine Ausnahme bildet die generalisierte pansklerotische Morphea.
-
Intensiv ausgeprägte Hautbeteiligung kann lebensbedrohlich sein, eine exakte Diagnose ist für eine Therapie notwendig.
-
Man muss Patientinnen und Patienten ausführlich untersuchen und dafür sind Hautbiopsien notwendig.
Auf dem Weg zur korrekten Diagnose seien Anamnese und Hautbiopsie wichtig, gegebenenfalls bildgebende Verfahren – und ein Algorithmus, so Prof. Dr. med. Thomas Krieg, Köln, bei seinem Vortrag am DGRh-Kongress. Zunächst sei zu klären, ob eine angeborene oder erworbene fibrosierende Erkrankung vorliegt. Man müsse also feststellen, wann die Erkrankung begonnen hat und ob es einen familiären Zusammenhang gibt. Danach sei zu beurteilen, ob sie lokalisiert oder systemisch ist. Zu den erblichen Erkrankungen gehörten unter anderem das Stiff-Skin-Syndrom sowie das Werner-Syndrom (Progerie).
Case Report: Stiff-Skin-Syndrom
Wie Krieg berichtete, stellten die Eltern kurz nach der Geburt fest, dass sich die Haut ihres Kindes verdickte und hart wurde. Die Patientin habe sich schliesslich kaum noch bewegen können, da sich die Fibrosen über die Gelenke hinweg ausgedehnt hatten. Zunächst wurde eine angeborene Sklerodermie diagnostiziert. Biopsie und Elektronenmikroskopie zeigten jedoch ein charakteristisches histologisches Bild: Tatsächlich war es zu exzessiven Kollagenablagerungen gekommen und die Struktur der Kollagenmoleküle war verändert. Diese Erkenntnis habe dann zur korrekten Diagnose Stiff-Skin-Syndrom (SSS) geführt, welches klinisch mit massiven Kontrakturen an Oberschenkeln und Gesäss und einer Verdickung der Faszien um das 3- bis 4-Fache verbunden sei. Die Haut sei massiv involviert, andere Organsysteme jedoch nicht. Die für diese angeborene Erkrankung verantwortliche Mutation wurde erst später identifiziert, sie betrifft ein Protein der extrazellulären Matrix: Fibrillin-1.
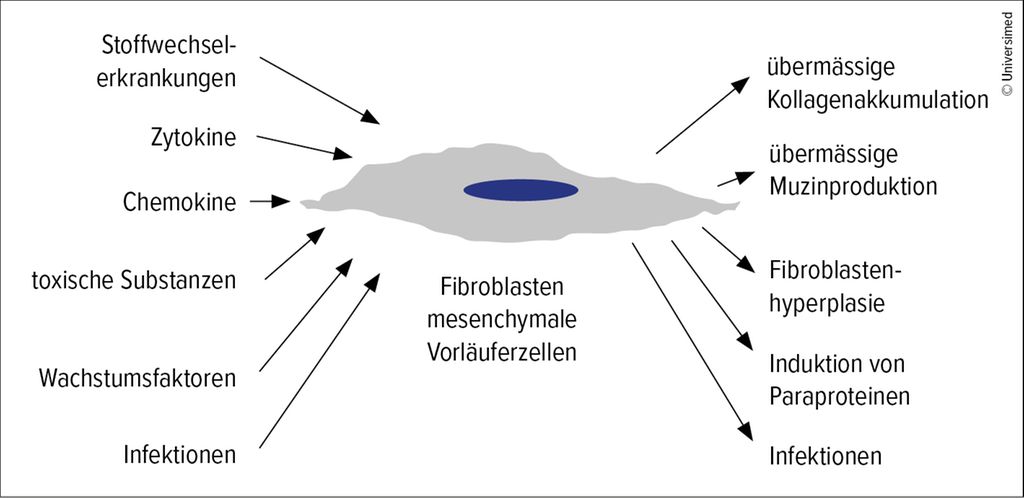
Abb. 1: Mechanismen bei fibrosierenden/sklerosierenden Hauterkrankungen
Verbindendes Element: der Wachstumsfaktor TGF-β
Der «transforming growth factor» β (TGF-β) spiele bei allen fibrosierenden Erkrankungen eine wesentliche Rolle, erklärte Krieg. Seine Aktivität werde über die Kopplung an ein «latent TGF-β binding protein» (LTBP) reguliert, das seinerseits an Fibrillin-1 binde. Eine Fibrillin-1-Mutation in der LTBP-Bindungsregion störe vermutlich die Regulation der TGF-β-Aktivität und induziere damit die Aktivierung der Fibrozyten. Je nachdem, wo diese Regulationsstörung erfolge, könne sie beispielsweise ein Stiff-Skin-Syndrom auslösen oder Marfan-ähnliche Symptome.
Erworben: die systemischen Sklerosen
Liege eine erworbene systemische Erkrankung mit antinukleären Antikörpern (ANA), Raynaud-Syndrom und mikrovaskulären Veränderungen vor, befinde man sich im Formenkomplex der systemischen Sklerosen, erklärte Krieg. Hier gebe es eine Bandbreite von Erkrankungen:
-
die limitierte kutane (lcSSc) sowie
-
die diffuse kutane (dcSSc) Variante,
-
Overlap-Syndrome wie das Polymyositis-Sklerodermie-Überlappungssyndrom (PM-Scl), die «mixed connective tissue disease» (MCTD) sowie Überlappungen mit der rheumatoiden Arthritis (SSc-RA) oder mit dem Lupus erythematodes (SSc-LE).
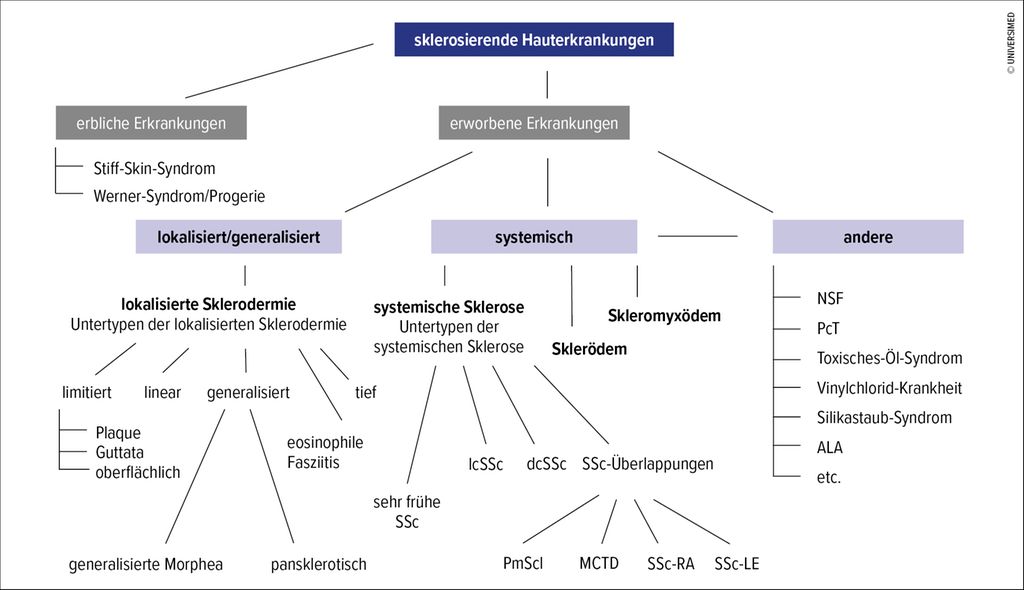
Abb. 2: Algorithmus zur Diagnose sklerosierender Hauterkrankungen (mod. nach Knobler R et al.)1, 2
Da alle eine Sklerosierung der Haut bedingten, seien zur Differenzierung entsprechende Laboruntersuchungen nötig. Zu beachten sei, dass sich die Krankheitsbilder interindividuell unterschiedlich entwickeln könnten, mahnte Krieg. Die Entwicklung von Frühformen mit geringer Krankheitslast und kaum verhärteter Haut, Raynaud-Syndrom und ANA, denen man in der Praxis oft begegne, sei daher schwer vorherzusehen. Es könne eine sehr frühzeitig aufgetretene systemische Sklerodermie (VEDOSS) vorliegen, die sich möglicherweise für viele Jahre oder Jahrzehnte kaum verändere. Doch könnten sich «Very early scleroderma»-Formen sehr progressiv entwickeln und in eine limitierte oder sogar diffuse SSc übergehen. Die Zusammenhänge würden noch untersucht.
Erworben, aber ohne Raynaud und ANA: Skleromyxödem und Scleroedema
Bei erworbenen Erkrankungen ohne Raynaud, ANA oder Veränderungen im Nagelfalz gebe es im Wesentlichen zwei für die Praxis relevante Erkrankungen, erklärte Krieg: das Skleromyxödem und das Scleroedema adultorum Buschke. Die Erkrankungen unterschieden sich erkennbar und liessen sich mit etwas Erfahrung bereits auf den ersten Blick diagnostizieren.
Die generalisierten papulären Hautveränderungen seien bei einem Skleromyxödem nicht sehr hart, eher wachsartig. Die Haut an den Händen, im Gesicht und im Mund sei besonders involviert, gerade im Gesicht sei die Faltenbildung deutlich zu erkennen. Das Syndrom sei üblicherweise mit einer monoklonalen Gammopathie assoziiert. Hier könnten ähnliche Ablagerungen wie in der Haut auch in Herz, Nieren, Lunge, Gastrointestinaltrakt, Nerven- und Muskuloskelettalsystem auftreten. Histologisch liessen sich Muzinablagerungen, Fibrose und eine Proliferation der Fibroblasten feststellen. Es handle sich um eine lebensbedrohliche Erkrankung, so Krieg.
Davon abgrenzen müsse man das Scleroedema adultorum Buschke. Hier lägen symmetrisch auftretende Indurationen vor, hauptsächlich in Nackenbereich und Gesicht, an den Schultern und den oberen Teilen des Integuments. Dies seien charakteristische Veränderungen. Histologisch sei die Epidermis unbeeinträchtigt, die Dermis jedoch massiv verdickt. Das Muzin fülle die Räume zwischen den Kollagenfasern aus. Im Gegensatz zum Skleromyxödem bestehe keine Proliferationssteigerung der Fibroblasten. Im Prinzip liessen sich drei Scleroedema-Typen unterscheiden:
-
assoziiert mit einem Diabetes
-
Erkrankung nach Streptokokkeninfektion
-
länger anhaltende Form, in der Regel assoziiert mit Paraproteinen
Lokalisierte Sklerodermien
Erkrankungen ohne systemische Beteiligung innerer Organe oder Raynaud-Syndrom, die sich lokal oder generalisiert auf der Haut manifestieren, gehörten zur Gruppe der lokalisierten Sklerodermien mit ebenfalls sehr unterschiedlichen Erkrankungsformen. Sie stellten wichtige Differenzialdiagnosen gegenüber systemischen Formen dar, so Krieg.
Nach europäisch vereinheitlichter Klassifikation werde zwischen limitierter, linearer, generalisierter Form und einem Mischtyp unterschieden. Ausserdem würden die eosinophile Fasziitis und die tiefe Morphea zu dieser Gruppe gezählt.
Bei dem im Bereich der limitierten Formen am häufigsten vorkommenden Plaque- Typ seien einzelne lokalisierte und zum Teil sehr ausgeprägte Plaques an Rumpf und Extremitäten typisch. Bleibe die Erkrankung auf wenige Stellen beschränkt, dann sei sie therapeutisch kein Problem. Jedoch könnten sich die Plaques schnell ausbreiten. Dabei sei im Zentrum ein sklerotischer Bereich zu beobachten, umrandet von einem charakteristischen lila Ring, der die fortschreitende Pathologie anzeigt. Dieser Typ sei therapeutisch hochproblematisch.
Zu den generalisierten Formen gehöre die schwer verlaufende generalisierte Sonderform, die «disabling pansclerotic morphea», die zwar auf die Haut beschränkt bleibe, sich jedoch mit dicken fibrotischen Plaques über das gesamte Integument ausdehnen und bis zur Einschränkung der Atmung führen könne. Sekundäre Phänomene, wie die Entwicklung von Plattenepithelkarzinomen, führten dann oft zum Tod. Es handle sich um eine schwerwiegende Erkrankung, die therapeutisch bis heute «eigentlich gar nicht» zu beherrschen sei.
Zu den häufigsten der linearen Formen zähle die Säbelhieb-Sklerodermie (Sklerodermie en coup de sabre) mit tiefen Läsionen, die meistens im subkutanen Fettgewebe und auch in den Knochen aufträten. Auch lineare Formen an den Extremitäten würden in der rheumatologischen Praxis häufig gesehen. Die Therapiebedürftigkeit dieser Formen sei deutlich höher als die isolierter Plaques irgendwo am Körper.
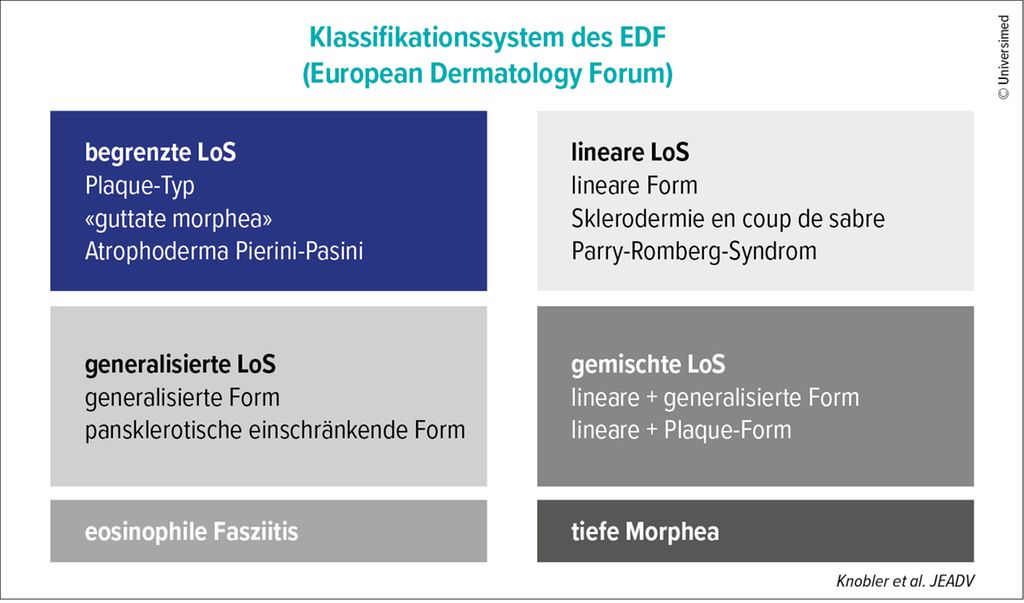
Abb. 3: Klassifikationssystem des European Dermatology Forum für lokalisierte Sklerodermien
Wichtig als Differenzialdiagnose zur systemischen Sklerose sei eine weitere generalisierte Sonderform: die eosinophile Fasziitis. Sie manifestiere sich hauptsächlich an den Extremitäten im tiefen subkutanen Gewebe und sei initial in der Regel mit einer peripheren eosinophilen Hypergammaglobulinämie verbunden, jedoch fehlten Raynaud-Syndrom, Autoantikörper und eine systemische Manifestation, sodass die Diagnose leicht zu stellen sei.
Andere erworbene Erkrankungen
Einige erworbene Krankheiten, so Krieg, liessen sich nicht in die aufgeführten klassischen sklerosierenden Erkrankungen einordnen. Auch hier litten Betroffene nicht am Raynaud-Syndrom, könnten aber Pseudosklerodermie-artige Hauterscheinungen haben. Dazu gehörten:
-
die Acrodermatitis chronica atrophicans nach Borrelien-Infektion,
-
die Graft-versus-Host-Reaktion in der sklerosierenden Variante,
-
heute seltene Erkrankungen wie die «nephrogenic systemic fibrosis» (NSF) sowie
-
die ebenfalls seltene Porphyria cutanea tarda, bei der man sich oft auf die sklerotischen Plaques konzentriere und vergesse, nach der Porphyrie zu suchen.
Die Erkrankungen seien wichtig, weil die Diagnostik schwierig sein könne und sie zum Teil übersehen würden. Ausserdem bräuchten sie andere Therapien und ein anderes Disease-Management. Sie zeigten darüber hinaus aber als Modelle, welche prinzipiellen Veränderungen zu systemischen fibrosierenden Erkrankungen führen und welche Rolle Eosinophile, mesenchymale Vorläuferzellen, Subgruppen von Fibroblasten sowie die Zytokinregulation spielten.
Biopsien, histologische Untersuchungen und Laboruntersuchungen
Biopsien und histologische Untersuchungen seien bei solchen Patientinnen und Patienten wichtig, um Differenzialdiagnosen abklären zu können, betonte Krieg. Sowohl Morbus Buschke als auch die NSF wiesen eine charakteristische Histologie auf. Und auch bei der Sklerodermie helfe die Histologie, die Diagnose zu bestätigen. Zum anderen sei die Entnahme einer Hautbiopsie hilfreich, um andere fibrosierende Erkrankungen auszuschliessen und um die Intensität der inflammatorischen Komponente histologisch zu bestimmen.
Bulk-Transkriptomanalysen könnten helfen, die Erkrankungsphase und -aktivität genauer einzugrenzen und unter Umständen eine molekulare Signatur zu gewinnen, um dann eventuell besser in klinische Studien eingreifen zu können. Anhand von «precision cuts» lasse sich herausfinden, ob die Erkrankung bei einem bestimmten Patienten oder einer bestimmten Patientin auf eine bestimmte Therapie anspricht. Und man könne mittels Biopsie und «spatial transcriptomics» die Expression kritischer Zytokine im Gewebe kartieren und damit die Pathogenese auf Zellebene beleuchten.
Die im individuellen Fall zielführenden labordiagnostischen Schritte sind in den Consensus-Statements zu Diagnostik und Therapie sklerosierender Hauterkrankungen enthalten.1,2
Quelle:
«Fibrotische Erkrankungen der Haut», Vortrag von Prof. Dr. med. Thomas Krieg, Köln, im Rahmen der Session «Fibrotische Erkrankungen – eine interdisziplinäre Herausforderung» am Deutschen Rheumatologie-Kongress, 19. September 2024, Düsseldorf
Literatur:
1 Knobler R et al.: Consensus statement on the diagnosis and treatment of sclerosing diseases of the skin, Part 1: Localized scleroderma, systemic sclerosis and overlap syndromes. J Eur Acad Dermatol Venereol 2024; 38(7): 1251-80 2 Knobler R et al.: Consensus statement on the diagnosis and treatment of sclerosing diseases of the skin, Part 2: Scleromyxoedema and scleroedema. J Eur Acad Dermatol Venereol 2024; 38(7): 1281-99
Das könnte Sie auch interessieren:

Tägliche Tablette gegen Psoriasis
Die US-Arzneimittelbehörde FDA hat mit Icotrokinra ein orales Medikament gegen Schuppenflechte zugelassen, welches die Rezeptoren für Interleukin-23 (IL-23) hemmt. Eine EU-Zulassung ...

Aminosäuren – Booster für die Wundheilung?
Für den Wundheilungsprozess ist je nach Heilungsprozess die richtige Kombination aus Kohlenhydraten, Fetten und Proteinen sowie aus Mineralien, Spurenelementen und Vitaminen essenziell. ...

Ein haariger Fall mit irreversiblen Folgen
Bestimmte Formen von Alopezie scheinen in jüngster Zeit explosionsartig zuzunehmen, wobei die genauen Ursachen bislang noch nicht vollständig geklärt sind. Handelt es sich dabei um eine ...