
Dermatologische Manifestationen von interstitiellen Lungenerkrankungen
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Interstitielle Lungenerkrankungen zählen zu den seltenen Erkrankungen und sind oft von einem schweren, progredienten und die Lebensqualität mindernden Verlauf geprägt. Extrapulmonale Manifestationen, wie beispielsweise dermatologische, sind ein wesentlicher Baustein in der Befunderstellung und Therapieabklärung
Interstitielle Lungenerkrankungen (ILD) führen durch Inflammation und/oder Vernarbung der alveolokapillären Membran zu Gasaustauschstörung und erhöhter Lungensteife und damit zu den Kardinalsymtomen Belastungsdyspnoe und Reizhusten.1 Die Pathogenese von ILD ist vielfältig und verschiedene Mechanismen können sich überlappen. Folgende ätiologische Einteilung hat sich bewährt:2
-
Idiopathische interstitielle Pneumonien (IIP): z.B. idiopathische Lungenfibrose (IPF) mit oft schlechter Prognose, aber auch entzündliche Formen wie die nicht spezifische interstitielle Pneumonie (NSIP) oder die kryptogen organisierende Pneumonie (COP)
-
Autoimmune ILD: z.B. mit Kollagenosen assoziierte ILD (CTD-ILD) oder rheumatoide Arthritis (RAILD)
-
Expositionsassoziierte ILD: z.B. exogen-allergische Alveolitis (EAA), Asbestose, ILD nach Radiotherapie
-
ILD mit Zysten oder Atemwegsausfüllung: meist seltene Formen wie die Lymphangioleiomyomatose oder die pulmonale Alveolarproteinose
-
Sarkoidose: eigene granulomatöse Systemerkrankung, die meist auch Lunge und thorakalen Lymphknoten betrifft
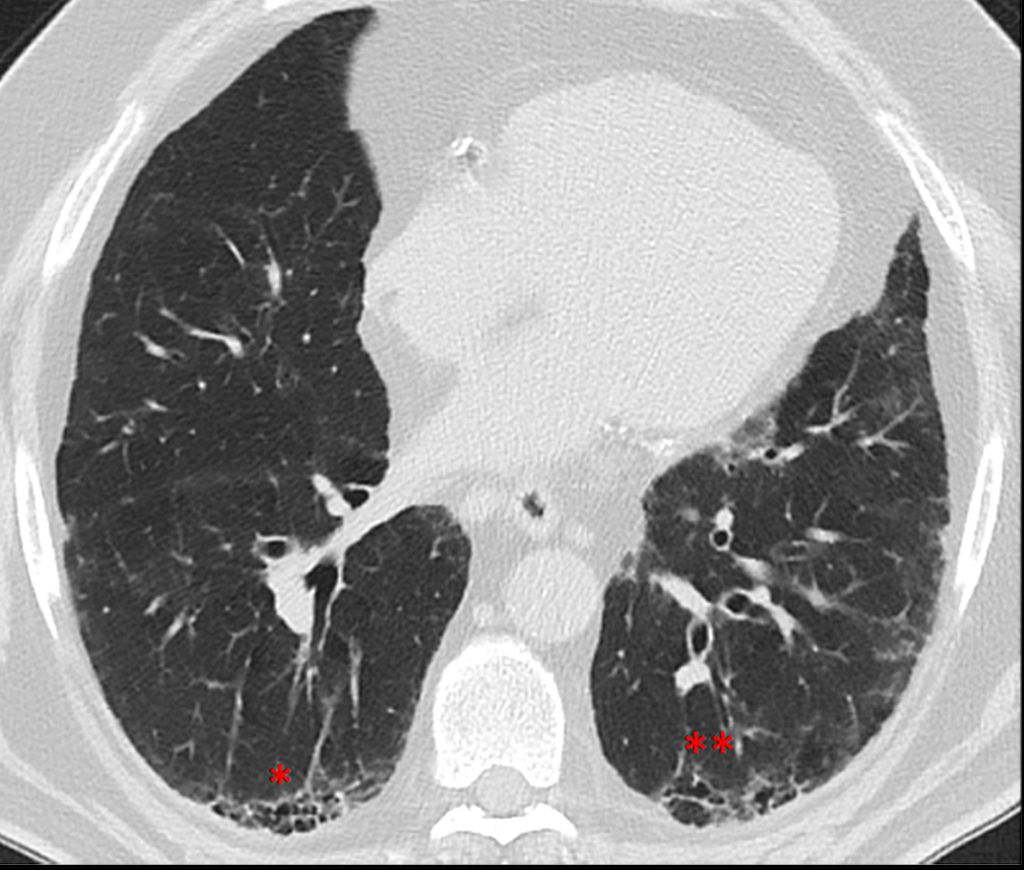
Abb. 1: Typisches UIP-Muster mit dorsobasalen Honigwabenzysten (* „honeycombing“) und peripher erweiterten Bronchi (** Traktionsbronchiektasen) bei einem 54-jährigen Patienten mit rheumatoider Arthritis
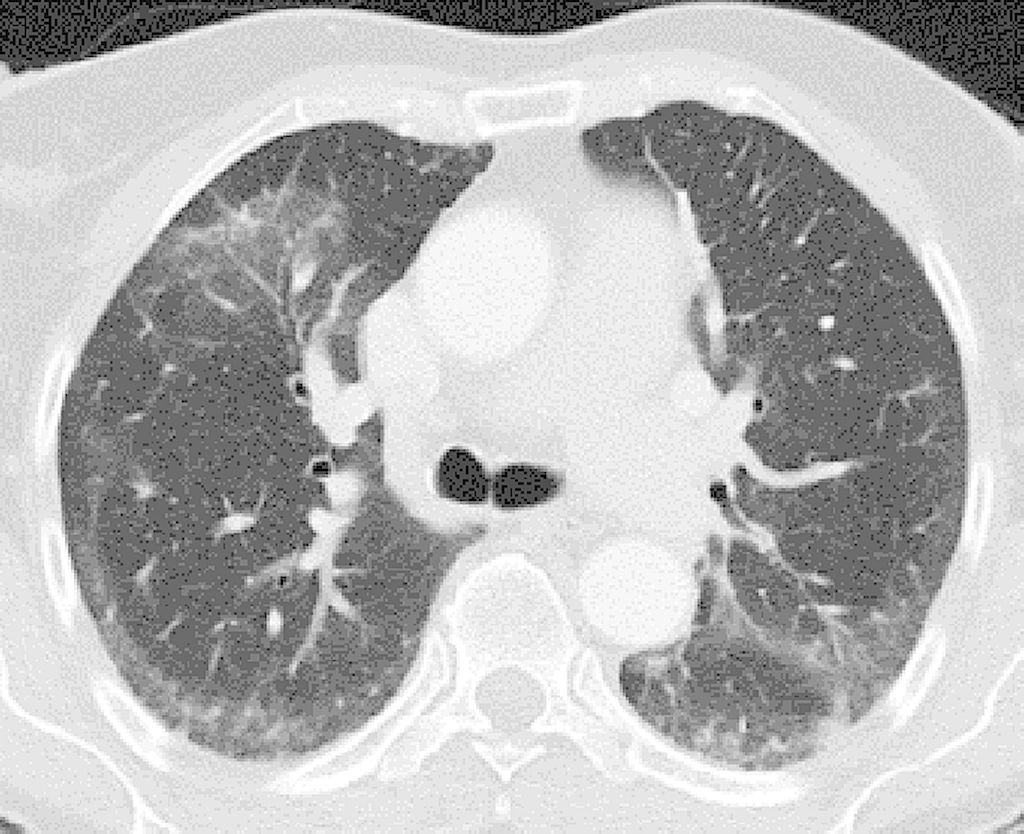
Abb. 2: Computertomografie-Bild eines klassischen NSIP-Musters mit subpleuralen Milchglasinfiltraten und Konsolidierungen
Verlauf und Prognose der verschiedenen ILD unterscheiden sich stark zwischen den Entitäten. Generell gesagt, sind idiopathische und progredient vernarbend verlaufende Formen wie die IPF prognostisch ungünstig und gehen mit einem medianen Überleben von nur 3–5 Jahren einher.3 Entzündlich mediierte Formen wie z.B. die NSIP oder die COP, oft im Rahmen von Autoimmunerkrankungen, oder die Sarkoidose haben meist eine bessere Prognose.4 Dies ist aber nicht nur von der Entität und Ursache, sondern z.B. auch vom radiologischen Muster abhängig, wobei ein „Usual interstitial pneumonia“(UIP)-Muster mit einer sehr ungünstigen Prognose einhergeht (Abb. 1).
Als seltene Erkrankungen sollten ILD an Zentren mit spezieller Expertise behandelt werden. Mittlerweile ist in der Diagnose und beim Therapieentscheid die multidisziplinäre Fallkonferenz (ILD-Board) von Pneumologie, Radiologie, Rheumatologie und Pathologie Standard.
Ein weiterer wichtiger Faktor in Diagnose und Therapie sind Begleiterkrankungen und extrapulmonale Manifestationen von ILD, die wesentlich zur Morbidität und Mortalität beitragen können.5 Dermatologische Manifestationen sind hier ein wesentlicher Befund, da sie einerseits auf eine zugrunde liegende Erkrankung schließen lassen können und andererseits bestehende Erkrankungen komplizieren können. Krankheitsspezifische Hautmanifestationen bei ILD-Patienten treten vor allem im Rahmen von Systemerkrankungen wie Autoimmunerkrankungen und auch bei der Sarkoidose häufig auf und diese Krankheiten sollen daher hier genauer abgehandelt werden.
Lungen- und Hautbeteiligung bei Autoimmunerkrankungen
Lungenbeteiligung tritt am häufigsten bei der systemischen Sklerose, bei Autoimmunmyositiden und bei rheumatoider Arthritis auf.4 Haut und Lunge gemeinsam sind am häufigsten bei systemischer Sklerose betroffen, sowohl bei der limitiert kutanen Form, häufiger aber bei der diffus kutanen Form (ILD bei über 50% der Patienten). Klassische dermatologische Manifestationen beinhalten digitale Ulzera, Sklerodaktylie und auch das Raynaud-Phänomen, während sich in der Lunge in den meisten Fällen das Bild einer nicht spezifischen interstitiellen Pneumonie (NSIP) manifestiert.6 Festzuhalten ist, dass ILD die häufigste Todesursache bei SSC ist und dass das Vorhandensein von ILD die Prognose der SSC wesentlich verschlechtert.6 Darum empfehlen aktuelle Leitlinien auch, dass schon bei Erstdiagnose einer SSC per CT auf das Vorhandensein einer ILD gescreent wird.7
Risikofaktoren für die Progression der ILD und für die pulmonal bedingte Mortalität sind eingeschränkte Lungenfunktion, ausgedehnte Lungenbeteiligung, männliches Geschlecht und Vorhandensein von SCL-70-Antikörpern.6,8 Speziell bei fortgeschrittener fibrotischer ILD ist auch eine pulmonale Krankheitsprogression ohne Fortschreiten anderer Organmanifestationen möglich.
Mehrere Therapieoptionen sind für die SSC verfügbar, die sowohl Wirkung auf die Haut und auch auf die Lunge zeigen. Standardtherapie ist aktuell meist Mycophenolat mofetil, bei rasch progredienten Verläufen wird auch Cyclophosphamid oder Rituximab verwendet.9,10 Zuletzt wurde auch die antifibrotisch wirkende Substanz Nintedanib speziell bei SSC-ILD zugelassen, die die Progression der ILD verlangsamen kann, Wirkung auf den Hautstatus zeigte sie indes nicht.11
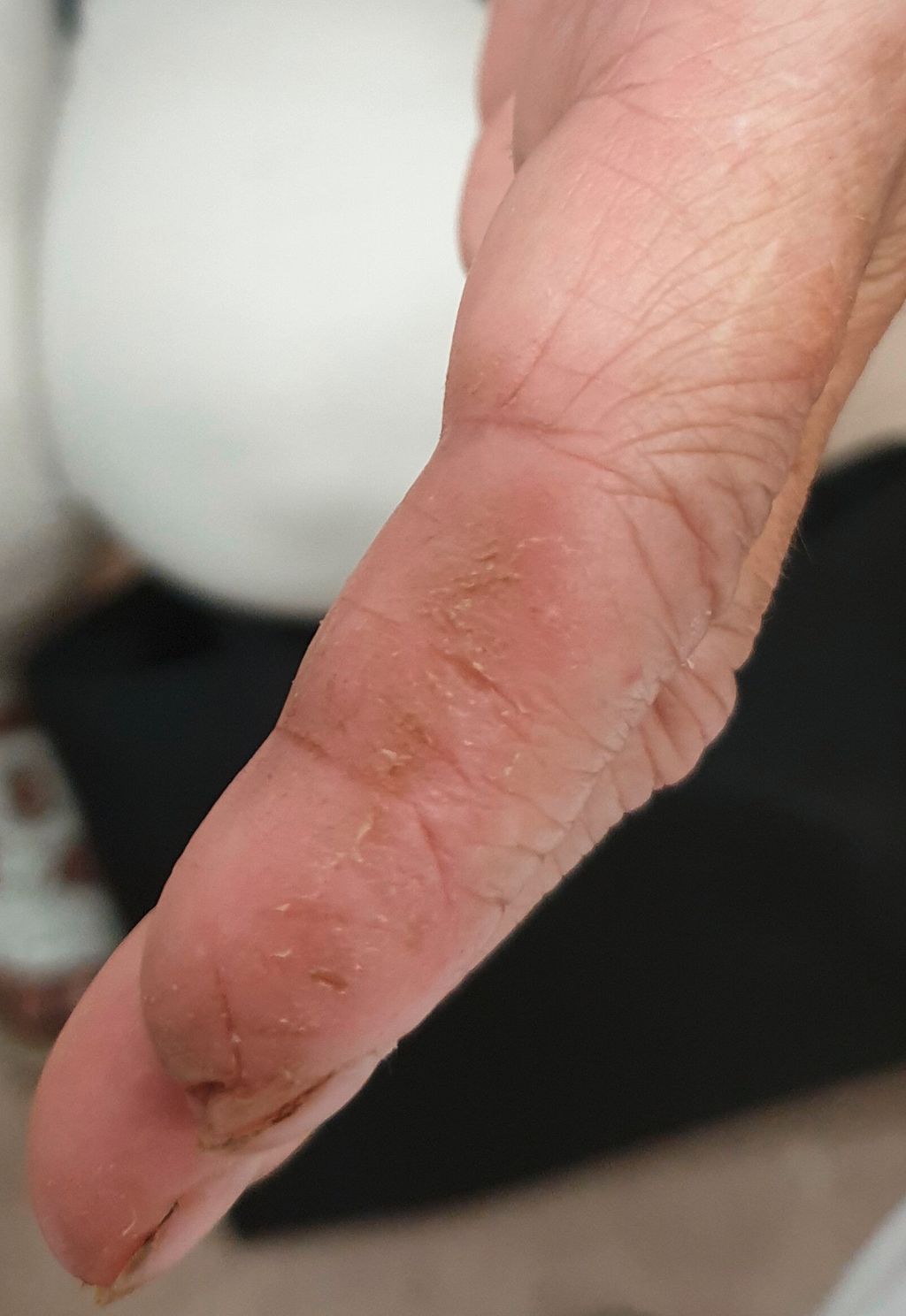
Abb. 3: Klassische „Mechanikerhände“ mit diffuser Schwellung und Ulzerationen bei einer 78-jährigen Patientin mit Jo-1-positivem „Antisynthetasesyndrom“
Eine weitere Krankheitsgruppe aus dem Formenkreis der Kollagenosen sind die Myositiden. Lungenbeteiligung ist hier häufig und oft mit spezifischen Phänotypen und Antikörperkonstellationen (z.B. Jo-1, PL-7 – „Antisynthetase-Antikörper“) assoziiert. Diese bestimmen oft auch die Prognose, wobei speziell MDA-5-Antikörper mit einer aggressiven Lungenbeteiligung und hoher Sterblichkeit einhergehen.12 Hautmanifestationen variieren ebenso stark bei verschiedenen Myositis-Subtypen, wobei bei Koinzidenz mit ILD häufig Grotton-Papeln oder sogenannte „Mechanikerhände“ vorliegen, wie in Abbildung 3 anhand eines Falles präsentiert. Zu beachten ist, dass bei Lungenmanifestation-Erkrankungen aus dem Myositis-Formenkreis zum Teil nur geringe oder auch fehlende klinische Symptomatik vorliegt („amyopathische“ Myositis).
Therapeutische Optionen bei Myositis mit Haut- und Lungenbeteiligung sind ähnlich wie bei systemischer Sklerose Kortikosteroide, Azathioprin, Cyclophosphamid oder Rituximab, wobei leider oft nur geringe Evidenz aus klinischen Studien vorliegt.4,12
Sarkoidose
Die Sarkoidose ist eine durch das Auftreten von nicht verkäsenden Riesenzellgranulomen gekennzeichnete Systemerkrankung, die meist Lunge und intrathorakale Lymphknoten betrifft, aber prinzipiell in jedem Organ auftreten kann.13 Die Ursache der Erkrankung ist weiterhin unklar, es wird von einem Zusammenspiel aus genetischer Disposition und externen Noxen ausgegangen.13
Die Haut ist mit bis zu 20% nach der Lunge einer der häufigsten Manifestationsorte.13,14 Typischerweise zeigen sich auch Hautläsionen in Form von Papeln, Knötchen oder Plaques. Klassisch ist auch das Auftreten im Bereich bestehender Narben oder Tätowierungen sowie im Gesicht als Lupus pernio. Histologisch kann aus diesen Läsionen der Nachweis von Granulomen erfolgen. Bei akuter Sarkoidose (Löfgren-Syndrom) kann sich auch ein Erythema nodosum als immunologisches Epiphänomen zeigen, das aber keine Granulome enthält. Die Sarkoidose an sich hat im Vergleich mit anderen ILD eine gute Prognose, systemische Therapieindikationen bestehen nur bei Organbedrohung oder signifikanten Beschwerden.15 Bei Hautsarkoidose kommt auch die kosmetische Relevanz der Läsionen als Therapieindikation hinzu. Therapie der Wahl bei Sarkoidose sind Glukokortikosteroide. Die Hautsarkoidose kann primär auch mit topischen Steroiden behandelt werden, jedoch zeigen diese bei ausgedehnteren Läsionen oft wenig Wirkung. Bleibt der Therapieerfolg unter Steroidtherapie aus oder kommt es zu Rezidiven, werden zusätzlich Immunmodulatoren wie Methotrexat oder als Drittlinientherapie TNF-alpha-Inhibitoren wie Infliximab verwendet.13,15
Fazit
Interstitielle Lungenerkrankungen sind oft schwere, progrediente und Lebensqualitäts-einschränkende Erkrankungen, die einer spezialisierten Abklärung und Therapie bedürfen. Insbesondere bei ILD im Rahmen von Systemerkrankungen können häufig auch Hautmanifestationen auftreten. Diese sind einerseits oft diagnostisch wegweisend, können aber andererseits auch zusätzliche Herausforderungen in der Therapie mit sich bringen.
Literatur:
1 Wijsenbeek M et al.: Interstitial lung diseases. Lancet 2022; 400: 769-86 2 Raghu G et al.: Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205: e18-e47 3 Simpson T et al.: The burden of progressive fibrotic interstitial lung disease across the UK. Eur Respir J 2021; 58: 2100221 4 Doyle TJ, Dellaripa PF: Lung manifestations in the rheumatic diseases. Chest 2017; 152: 1283-95 5 Jovanovic DM et al.: Comorbidity burden and survival in patients with idiopathic pulmonary fibrosis: the EMPIRE registry study. Respir Res 2022; 23: 135 6 Volkmann ER et al.: Systemic sclerosis. Lancet 2022; S0140-6736(22)01692-0 7 Hoffmann-Vold A-M et al.: The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol 2020; 2:e71-e83 8 Goh NSL et al.: Interstitial lung disease in systemic sclerosis. Am J Respir Crit Care Med 2008; 177: 1248-54 9 Tashkin DP et al.: Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016; 4: 708-19 10 Maher TM et al.: Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med 2023; 11: 45-54 11 Distler O et al.: Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019; 380: 2518-28 12 Hallowell RW, Paik JJ: Myositis-associated interstitial lung disease: a comprehensive approach to diagnosis and management. Clin Exp Rheumatol 2022; 40: 373-83 13 Valeyre D et al.: Sarcoidosis. Lancet 2014; 383: 1155-67 14 Schupp JC et al.: Phenotypes of organ involvement in sarcoidosis. Eur Respir J 2018; 51(1): 1700991 15 Baughman RP et al.: ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J 2021; 58: 2004079
Das könnte Sie auch interessieren:
Behandlungspfad „Atopische Dermatitis“
Das Netzwerk onkoderm hat seine Empfehlungen für die ambulante Versorgung von Patienten mit atopischer Dermatitis aktualisiert: Der onkoderm-Behandlungspfad 2025 sowie weitere Tools ...

Perianale Dermatosen
Perianale Dermatosen sind ein klinisch vielgestaltiges und herausforderndes Krankheitsbild, das sowohl dermatologische als auch proktologische Kompetenzen erfordert. Dieser Überblick ...

Wege zur korrekten Diagnose
Patientinnen und Patienten mit verhärteter Haut können an sehr unterschiedlichen Erkrankungen leiden. Im Zentrum steht immer ein Fibroblast, eine mesenchymale Vorläuferzelle, die durch ...