Wie lässt sich die Zeit bis zur Diagnose der amyotrophen Lateralsklerose verkürzen?
Die amyotrophe Lateralsklerose (ALS) gilt als unaufhaltsam fortschreitende neurodegenerative Erkrankung, die im Durchscnitt drei bis vier Jahre nach Symptombeginn mit dem Tod durch Versagen der Atemwegsmuskulatur einhergeht. Ein frühzeitiger Nachweis der Erkrankung ist aus medizinischer und psychosozialer Sicht von essenzieller Bedeutung, um unnötige Eingriffe zu verhindern und Patient:innen einen früheren Eintritt in klinische Studien und multidisziplinäre Behandlungseinrichtungen zu ermöglichen.1
Obwohl die Veröffentlichung der Gold-Coast-Kriterien im Jahr 2020 und die Implementierung der Elektromyografie (EMG) die Diagnose der ALS in der klinischen Praxis erheblich vereinfachten, blieb die Zeit bis zur Diagnosestellung in den letzten zwei Dekaden nahezu unverändert.1–3 So zeigten Studien bei mehr als 2500 Patient:innen aus überwiegend europäischen Zentren, dass zwischen dem Auftreten der ersten Symptome und der ALS-Diagnose häufig 11 bis 18 Monate vergehen.4,5
Faktoren, welche die Diagnose verzögern
Die Gründe für diese Verzögerung sind bis dato noch nicht eindeutig geklärt. Ein höheres Alter, die Lokalisation der ersten Symptome (spinal vs. bulbär) sowie eine Überweisung zu nichtspezialisierten Fachärzt:innen der Neurologie gegenüber ALS-Spezialist:innen scheinen wesentliche Einflusskriterien für eine verzögerte Diagnose zu sein. Besonders in frühen Stadien der Erkrankung, wenn beispielsweise weniger als zwei Körperstellen betroffen sind, bei atypischem Phänotyp oder bei Komorbiditäten eignen sich die Gold-Coast-Kriterien nur eingeschränkt für die Diagnose durch Nicht-Spezialist:innen. Zudem erfüllt etwa ein Fünftel der ALS-Patient:innen in frühen Stadien die notwendigen EMG-Kriterien nicht.1
Instrumente für die Beschleunigung der Diagnose
Ultraschallmessungen könnten in der Zukunft einen wesentlichen Beitrag zur Früherkennung der ALS leisten. So zeigte der Nachweis von Faszikulationen in mindestens drei Muskelgruppen, einem frühen Erscheinungsbild der ALS, eine Sensitivität von 89% und eine Spezifität von 88% für die Unterscheidung von ALS und ALS-ähnlichen Erkrankungen.6
Ausserdem rückt das Interesse an fluiden Biomarkern zunehmend in den Vordergrund. Neurodegenerative Erkrankungen wie die Alzheimer- oder die Parkinsonkrankheit legen den Verdacht nahe, dass auch ALS mit einer Erhöhung der Plasmaspiegel von Neurofilament-Leichtketten (NfL) während einer präsymptomatischen Phase einhergeht (Abb 1). Vor allem bei Trägern einer ALS-Mutation könnte deren Nachweis somit eine wesentliche Beschleunigung der Diagnosestellung ermöglichen.1 Andere interessante diagnostische Biomarker, wie beispielsweise das TAR-DNA-bindende Protein (TDP-43), die Superoxid-Dismutase 1 (SOD1) oder das Fused-in-Sarcoma(FUS)-Protein sind Gegenstand der aktuellen Forschung für die Früherkennung der ALS.1
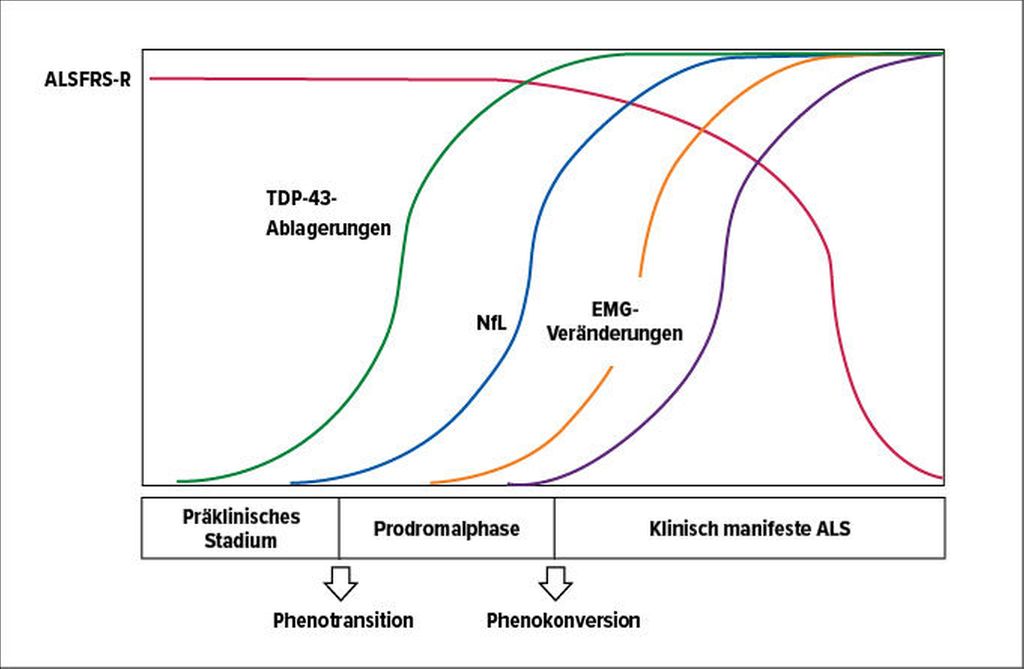
Abb. 1: Hypothetisches Szenario eines kaskadenartigen Verlaufs der ALS. Phase 1: präklinisches Stadium, in dem erhöhte Plasmakonzentrationen von NfL nachgewiesen werden können. Phase 2: Prodromalphase mit unspezifischen Symptomen und ersten Veränderungen in der EMG. Phase 3: klinisch manifeste Phase mit eindeutigen Symptomen der ALS (modifiziert nach García-Casanova PH et al.)1
Fazit
Die frühe Diagnose der ALS stellt trotz wesentlicher technischer und wissenschaftlicher Fortschritte eine enorme Herausforderung dar. Während Biomarker wie NfL und moderne Bildgebungsverfahren interessante Anhaltspunkte für weitere Forschung bieten, verbleibt die rasche Überweisung an spezialisierte ALS-Zentren bei frühen Warnsignalen die effektivste Massnahme zur Diagnose in frühen Erkrankungsstadien.
Quelle:
García-Casanova P et al.: Advances in the early diagnosis of amyotrophic lateral sclerosis. Expert Rev Neurother 2025; 25(4): 415-25
Literatur:
1 García-Casanova PH et al.: Advances in the early diagnosis of amyotrophic lateral sclerosis. Expert Rev Neurother 2025; 25(4): 415-25 2 Turner MR et al.: Diagnosing ALS: the Gold Coast criteria and the role of EMG. Pract Neurol 2022; 22(3): 176-8 3 Sanghani N et al.: Electrodiagnostic findings in amyotrophic lateral sclerosis: variation with region of onset and utility of thoracic paraspinal muscle examination. Muscle Nerve 2024; 69(2): 172-78 4 Goyal NA et al.: Misdiagnosis of amyotrophic lateral sclerosis in clinical practice in Europe and the USA: a patient chart review and physician survey. Amyotroph Lateral Scler Frontotemporal Degener 2024; 25(1-2): 16-25 5 Falcão de Campos C et al.: Trends in the diagnostic delay and pathway for amyotrophic lateral sclerosis patients across different countries. Front Neurol 2023; 13: 1064619 6 Hannaford AP et al.: Muscle ultrasound aids diagnosis in amyotrophic lateral sclerosis. Clin Neurophysiol 2025; 170: 234-43
Das könnte Sie auch interessieren:

Diagnose der ALS: von Biomarkern bis Kognition und Verhalten
Die Heterogenität der ALS macht eine Diagnose nicht leicht. Dazu kommt, dass die Pathogenese immer noch nicht richtig verstanden ist und dass sich die Frühsymptome mit jenen anderer ...

Ist die ketogene Diät eine Präzisionsmedizin?
Die ketogenen Ernährungstherapien sind etablierte Behandlungsformen bei Epilepsie. Während sie primär bei therapierefraktären pädiatrischen Epilepsien eingesetzt werden, finden sie ...

Neues aus der Alzheimer’s Disease Drug Development Pipeline
Mit der weltweiten Zulassung der Amyloidantikörper Lecanemab und Donanemab ist erstmals eine kausale Behandlung der Alzheimerkrankheit möglich geworden. Die Behandlung setzt an der ...