Mehr als nur eine trockene Angelegenheit: Die Sjögren-Erkrankung im Fokus
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Sjögren-Erkrankung (SjD) ist eine chronisch-entzündliche, systemische Autoimmunerkrankung mit hoher klinischer Variabilität. Sie gehört zur Familie der Kollagenosen, zu der beispielsweise auch die systemische Sklerose, der systemische Lupus erythematodes oder die Polymyositis gehören. Dieser Übersichtsartikel gibt einen aktuellen Überblick über Klinik, Diagnostik und Therapie und will internistisch tätige Ärzt:innen für die differenzierte Versorgung dieser oft unterschätzten Erkrankung sensibilisieren.
Charakteristisch für die SjD ist eine lymphozytäre Infiltration exokriner Drüsen, insbesondere der Speichel- und Tränendrüsen, was zu deren Funktionsbeeinträchtigung führt. Folge ist die typische orale respektive okuläre Sicca-Symptomatik. Die Trockenheitsproblematik kann aber auch Nase, Rachen, Haut oder Vulva/Vagina betreffen. Darüber hinaus kommt es bei rund einem Drittel der Betroffenen zu einem extraglandulären Befall, wobei praktisch jedes Organsystem betroffen sein kann.
Terminologie
Benannt ist die Erkrankung nach dem schwedischen Augenarzt Henrik Sjögren, welcher in seiner 1933 auf Deutsch publizierten Dissertation eine Keratoconjunctivitis sicca bei 19 Frauen beschrieben hat, von denen 13 auch Gelenkmanifestationen aufwiesen.1 International hat sich in den letzten Jahren auf Empfehlung von Sjögren-Expert:innen und Betroffenen der Terminus «Sjögren-Erkrankung» (englisch «Sjögren’s disease») gegen den früheren Begriff «Sjögren-Syndrom» durchgesetzt. Damit wird dem Umstand Rechnung getragen, dass es sich um eine ernst zu nehmende Systemerkrankung handelt und nicht um eine Aneinanderreihung von Symptomen, wie es der Begriff «Syndrom» suggerieren könnte.
Die SjD kann als alleinstehende Erkrankung bestehen («primäre Sjögren-Erkrankung») oder zusammen mit einer anderen chronisch-entzündlichen Autoimmunerkrankung auftreten, z.B. einer rheumatoiden Arthritis oder einem systemischen Lupus erythematodes. Was im zweiten Fall früher als «sekundäres Sjögren-Syndrom» bezeichnet wurde, nennt sich heute «assoziierte Sjögren-Erkrankung».
Pathogenese und Ätiologie
Die Pathogenese der SjD ist komplex und nicht vollständig verstanden. Eine zentrale Rolle spielen genetische Prädispositionen, Umweltfaktoren sowie hormonelle Faktoren.
Als Umweltfaktoren werden insbesondere Infektionen mit Epstein-Barr-Virus (EBV) und Zytomegalievirus (CMV) angenommen.2 Zudem scheint der mit der Menopause einhergende Östrogenmangel die Krankheitsentstehung zu triggern, was den Umstand erklären könnte, dass die Erkrankung häufig bei postmenopausalen Frauen diagnostiziert wird.3 Bei der SjD zeigt sich der autoimmune Prozess vor allem durch die Schädigung exokriner Drüsen, die Bildung von Autoantikörpern, die Ablagerung von Immunkomplexen sowie durch die Infiltration verschiedener Organe mit Lymphozyten im Rahmen eines systemischen Verlaufs. Eine zentrale Rolle spielen dabei eine gesteigerte Produktion des B-Zell-aktivierenden Faktors (BAFF) sowie eine übermässige Aktivierung der B-Zellen, bedingt durch eine veränderte Expression des BAFF-Rezeptors in unterschiedlichen Immunzelltypen.4
Klinisches Bild
Leitsymptom der SjD ist die Sicca-Symptomatik. Die Augentrockenheit kann sich mit brennenden, juckenden Augen, Sandgefühl und Notwendigkeit von befeuchtenden Augentropfen äussern. Manche Betroffene spüren überhaupt keine Augentrockenheit, obwohl sie in quantitativen und qualitativen Messungen nachweisbar ist.
Die Mundtrockenheit ist häufig nachts betont, kann zu Veränderung des Geschmacksinnes und zu Schwierigkeiten beim Schlucken von trockenen Speisen wie z.B. Brot führen. Folgen des Speichelmangels sind unter anderem ein erhöhtes Risiko für Karies und Mundsoor.5
Bei einigen Betroffenen kommt es zu rezidivierenden, häufig selbstlimitierenden Schwellungen der Speicheldrüsen.
Viele SjD-Betroffene klagen zudem über Myalgien und Arthralgien, die oft klinisch nicht als klare Myositis oder Arthritis fassbar sind und manchmal dem Bild eines fibromyalgischen Syndroms ähneln.6
Bei bis zu 70% aller SjD-Betroffenen kommt es auch zu einer chronischen Fatigue, wobei ein direkter Zusammenhang mit proinflammatorischen Signalwegen nicht nachgewiesen werden konnte.7
Extraglanduläre Manifestationen sind vielfältig. Nicht selten wird die SjD erst im Rahmen der ätiologischen Abklärung einer unklaren Polyneuropathie, interstitiellen Lungenerkrankung, tubulointerstitiellen Nephritis oder Glomerulonephritis diagnostiziert. In seltenen Fällen können die für das SjD typischen SSA/Ro-Autoantikörper ab der 16. Schwangerschaftswoche zu einem kongenitalen AV-Block führen. Auch dieser kann Anlass für weiterführende Abklärungen sein, in deren Verlauf letztlich erst die Diagnose einer SjD gestellt wird.
Ferner kann die SjD zu Arthritiden führen, die typischerweise anerosiv verlaufen und häufig symmetrisch MCP-, PIP- und Handgelenke betreffen. Der Hautbefall reicht von vaskulitischen Manifestationen wie Petechien und Purpura bis hin zu anulärem Erythem oder Erythema-multiforme-ähnlichen Läsionen. Manche haben ein kältegetriggertes Raynaud-Phänomen, das häufig milde ausgeprägt ist und im Gegensatz zur systemischen Sklerose typischerweise keine trophischen Störungen wie Ulzera oder «pitting scars» zur Folge hat. Die Kapillarmikroskopie zeigt in der Regel einen Normalbefund oder unspezifische Veränderungen wie eine verminderte Kapillardichte, aber keine organische Mikroangiopathie.8
Abbildung 1 zeigt eine Übersicht über die häufigsten systemischen Manifestationen bei der SjD.
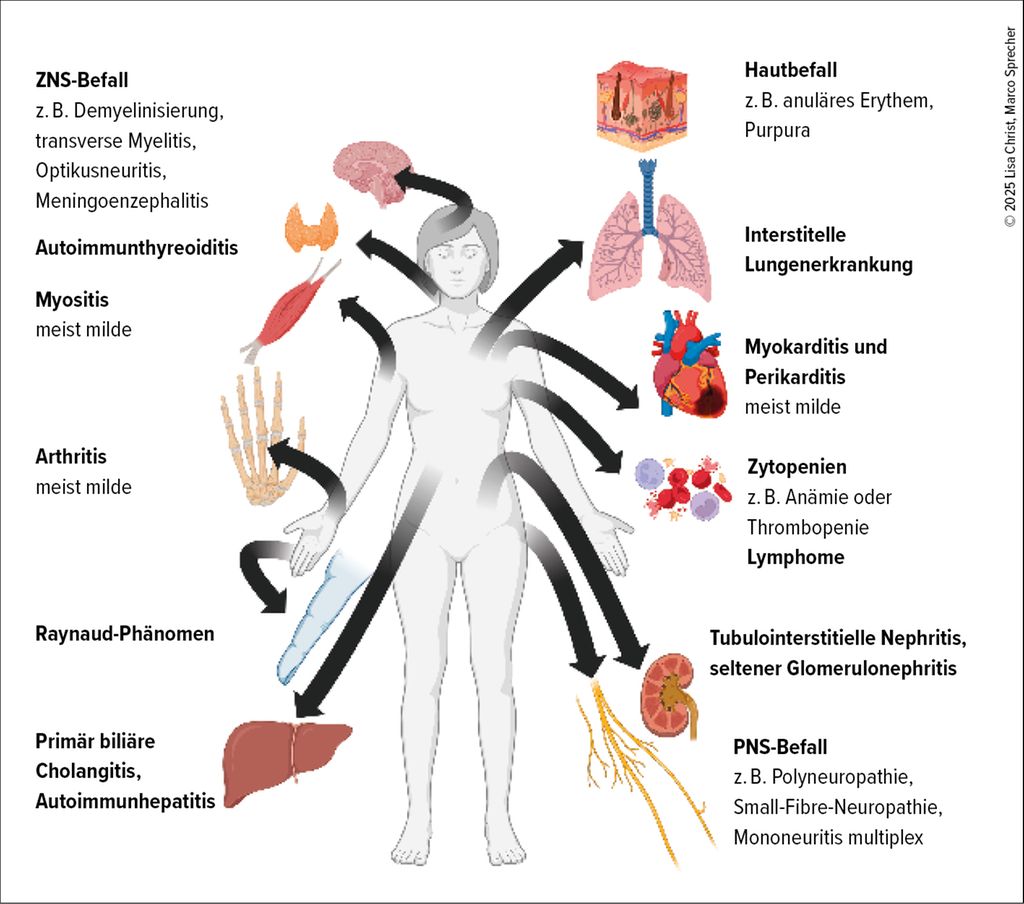
Abb. 1: Übersicht über die häufigsten extraglandulären Manifestationen bei der Sjögren-Erkrankung (mit © Biorender erstellte Grafik)
Diagnostik
Die Diagnose basiert auf einer Kombination aus klinischer Beurteilung, laboranalytischen Befunden und funktionellen Tests. Die aktuell gültigen Klassifikationskriterien der europäischen und der amerikanischen Rheumaliga (EULAR resp. ACR) beinhalten fünf Domänen, denen jeweils eine unterschiedliche Anzahl an Punkten zugeordnet werden (Tab.1). Ein Wert ≥4 Punkte erlaubt die Klassifikation als SjD.
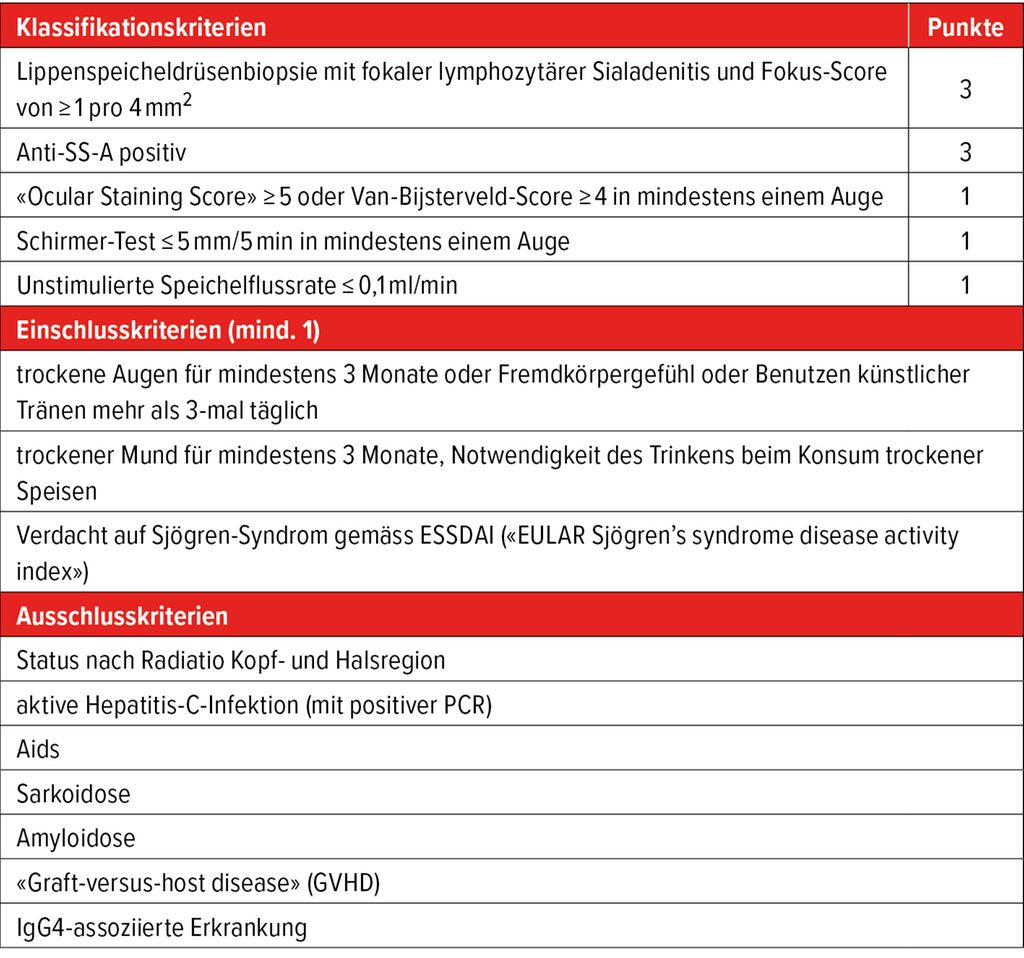
Tab. 1: ACR/EULAR-Klassifikationskriterien für die Sjögren-Erkrankung 2016. Die Klassifikationskriterien sind erfüllt, wenn mindestens 4 Punkte, mindestens 1 Einschlusskriterium und kein Ausschlusskriterium erreicht werden
Typische serologische Marker sind antinukleäre Antikörper (ANA), Anti-Ro/SSA- und Anti-La/SSB-Antikörper, Rheumafaktoren sowie eine polyklonale Hypergammaglobulinämie, in manchen Fällen auch Kryoglobuline. Rund 60% der SjD-Betroffenen haben positive Rheumafaktoren, was eine höhere B-Zell-Aktivität widerspiegelt und oft mit einer höheren systemischen Krankheitsaktivität einhergeht.9 Circa 15% der SjD-Betroffenen sind negativ für ANA, Anti-Ro/SSA-, Anti-LA/SSB-Antikörper und Rheumafaktoren.10
Funktionelle Tests wie der Schirmertest und die Sialometrie sind wichtige Parameter zur Objektivierung der Sicca-Symptomatik. Beim Schirmertest wird ein standardisierter Papierfilterstreifen in das Unterlid gehängt und nach fünf Minuten die Länge des durch Tränenflüssigkeit befeuchteten Papieranteils gemessen. Ein Wert von 5mm oder weniger gilt als pathologisch. Bei der unstimulierten Sialometrie wird der Speichel im Sitzen ohne äussere Reize über einen definierten Zeitraum (meist 5–15 Minuten) gesammelt durch freies Ausspucken in ein Messgefäss. Die stimulierte Sialometrie erfolgt unter kontrollierter Reizung, etwa durch Kauen auf standardisiertem Paraffinwachs oder Kaugummi, wobei der produzierte Speichel ebenfalls in einem bestimmten Zeitfenster gesammelt und gemessen wird. Häufig wird eine unstimulierte Sialometrie von ≤0,1ml/min als pathologisch erachtet, was jedoch umstritten ist, da dieser Wert alters- und geschlechtsspezifische physiologische Schwankungen nicht berücksichtigt und eine sehr niedrige Sensitivität hat (ca. 50%) im Gegensatz zum Schirmertest (ca. 70%). Manche Expert:innen empfehlen daher einen Schwellenwert von ≤0,2ml/min.11

Abb. 2: Sonografie der Parotisdrüse, Grad 2 nach OMERACT: Inhomogenitäten der Parotistextur mit multiplen hypo- bis anechogenen Arealen, welche noch von normaler Gewebetextur ummantelt sind
Bildgebende Verfahren wie die Sonografie der Parotis- und Submandibularisdrüsen haben in den letzten Jahren an Bedeutung zugenommen, da sie sensitiv, nichtinvasiv und schnell durchführbar sind. Im B-Bild lassen sich mit einer linearen 10- bis 18-MHz-Probe unterschiedlich ausgeprägte Inhomogenitäten des Speicheldrüsenparenchyms feststellen, mit fokalen hypo- bis anechogenen rundlichen Arealen (Abb. 2). Breite internationale Akzeptanz hat die Einteilung dieser Veränderungen in verschiedene Grade (0–3) nach OMERACT («Outcome Measures in Rheumatology Clinical Trials») erlangt.12 Sonografisch werden die Speicheldrüsen mit dem Parenchym der Schilddrüse verglichen, welche sich im Normalfall homogen und zueinander isoechogen zeigen sollten. Zusätzlich kann auch nach OMERACT die Hypervaskularisierung mittels Farbdoppler in verschiedene Gradeinteilungen (0–3) quantifiziert werden.13 Auf Essen und Kauen sollte mindestens eine Stunde vorher verzichtet werden, da der Kauprozess physiologischerweise die Durchblutung anregt.
Eine systematische Übersichtsarbeit mit Metaanalyse ergab, dass die Sonografie der Speicheldrüsen bei der Diagnostik eine gepoolte Sensitivität von 80% (95%-Konfidenzintervall [95%CI]: 77–83%) sowie eine Spezifität von 90% (95% CI: 87–92%) aufweist.14 In einer anderen Studie wurde zudem eine hohe Übereinstimmung der Ultraschallbefunde mit Biopsieergebnissen der Ohrspeicheldrüse (83%) bzw. der Unterlippe (79%) festgestellt. Besonders aussagekräftig war in ihrer Untersuchung die Kombination aus einem pathologischen Sonografiebefund mit dem Nachweis von Ro/SSA-Autoantikörpern bzw. einem unauffälligen Ultraschallbild bei gleichzeitigem Fehlen dieser Antikörper – beide Konstellationen wiesen eine hohe prädiktive Aussagekraft für die Zuordnung zur Sjögren-Diagnose oder deren Ausschluss auf.15
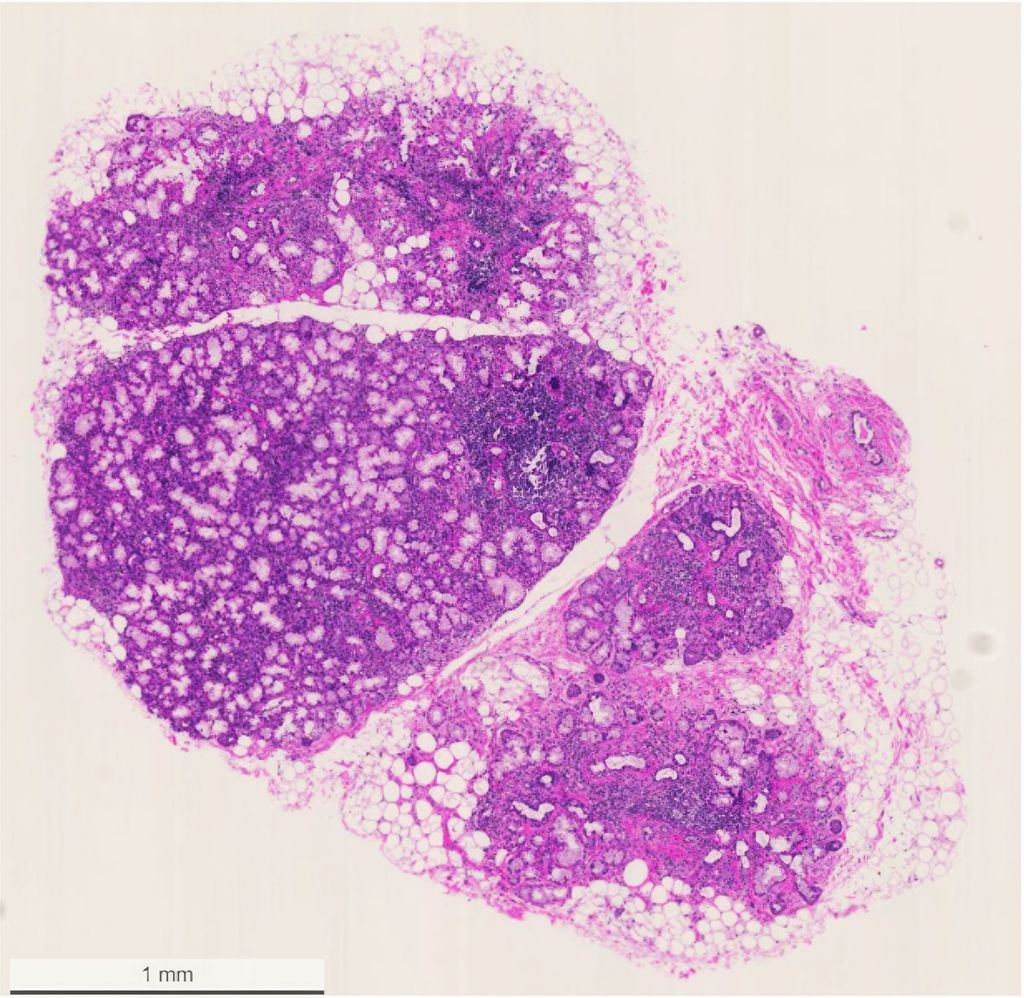
Abb. 3: Biopsie der Speicheldrüse an der Unterlippe mit Nachweis multipler lymphozytärer Foci (mit freundlicher Genehmigung der Patientin)
Die Speicheldrüsenbiopsie bleibt trotzdem ein zentraler Bestandteil der Diagnostik, ist jedoch nicht zwingend erforderlich, wenn das übrige klinische Bild eindeutig ist. Unter lokaler Betäubung werden dabei mehrere Speicheldrüsenläppchen, in der Regel aus der Innenseite der Unterlippe, entnommen – unter sorgfältiger Schonung von Gefässen und Nerven. Charakteristisch für die SjD ist der histologische Nachweis lymphozytärer Foci – fokale, meist periduktale Infiltrate aus mindestens 50 Lymphozyten und Histiozyten (Abb.3). Der sogenannte Focus-Score beschreibt die Anzahl dieser lymphozytären Foci pro 4mm2 Drüsengewebe. Ein Wert ≥1 gilt als diagnostisch wegweisend für die SjD, ist aber nicht pathognomonisch. Beispielweise kann eine unbehandelte HIV-Infektion in den Speicheldrüsen ein sehr ähnliches histologisches Bild zeigen.16 Die Speicheldrüsenbiopsie leistet andererseits einen wichtigen Beitrag zur Differenzialdiagnostik – etwa zur Abgrenzung gegenüber IgG4-assoziierten Erkrankungen, Sarkoidose, Amyloidose oder malignen Prozessen, welche ebenfalls eine Ursache für die Sicca-Symptomatik sein können. Der Nachweis ektoper Keimzentren ist zudem prognostisch relevant, da sich das Lymphomrisiko – je nach Studie – um das bis zu 7,8-Fache erhöhen kann.17
Lymphomrisiko
Ein besonderes Augenmerk verdient das erhöhte Risiko für die Entwicklung eines B-Zell-Non-Hodgkin-Lymphoms, das bei etwa 5–10% der SjD-Betroffenen eintritt.18,19 Risikofaktoren sind u.a. persistierende Parotisschwellung, Hypokomplementämie, Kryoglobulinämie und monoklonale Gammopathie.20 Seronegative SjD-Betroffene haben hingegen ein niedrigeres Lymphomrisiko, es liegt je nach Studie bei ca. 1% (Quartuccio et al., 2015),18 ist aber immer noch höher als in der Normalbevölkerung.
Histologisch handelt es sich meist um extranodale Marginalzonenlymphome (MALT-Lymphome), die sich bevorzugt in den Speicheldrüsen oder anderen mukosalen Geweben manifestieren. Klinisch können sie sich durch Lymphadenopathie, Organomegalie oder systemische B-Symptomatik äussern. Die Überwachung von Risikopersonen erfolgt durch regelmässige klinische Untersuchungen, serologische Verlaufsparameter (z.B. Blutbild, Komplement, Beta-2-Mikroglobulin, CD4/8-Verhältnis) und ggf. bildgebende Verfahren. Einen international akzeptierten Konsens über das genaue Monitoring gibt es bis dato nicht. Eine frühzeitige Diagnostik und interdisziplinäre Betreuung sind aber entscheidend für eine gute Prognose.
Therapie
Die Therapie der SjD orientiert sich an der Symptomatik, dem Schweregrad der Erkrankung sowie der Art und Ausprägung der Organbeteiligung. Zwar gibt es Therapieempfehlungen wie von der europäischen Rheumaliga EULAR (European League Against Rheumatism),21 ein evidenzbasiertes, einheitliches Therapieschema existiert bislang aber nicht, weshalb ein individualisierter, multidisziplinärer Ansatz empfohlen wird.
Behandlung der Sicca-Symptomatik
Die symptomatische Behandlung der Sicca-Symptomatik stellt die Basistherapie dar und sollte individuell angepasst sowie regelmässig evaluiert werden.
Okuläre Sicca-Symptomatik
Erste Wahl sind künstliche Tränenersatzmittel. Dazu gibt es Augentropfen mit diversen Inhaltsstoffen auf dem Markt, z.B. Hyaluronsäure, Carbomere, Hypromellose, Povidon K25 oder Carmellose. Sind sehr häufige Applikationen pro Tag notwendig, sollten bevorzugt konservierungsmittelfreie Präparate verwendet werden, da Konservierungsmittel bei häufiger Anwendung die Augen zusätzlich reizen können. Visköse Augengele oder Vitamin-A-haltige Augensalben werden insbesondere für die Nacht empfohlen, da sie eine lang anhaltende Befeuchtung der Augenoberfläche ermöglichen, jedoch aufgrund der Konsistenz zu vorübergehendem Schleiersehen führen können. Lipidhaltige Präparate wiederum führen dazu, dass der Tränenfilm weniger schnell aufbricht oder verdunstet. Sie kompensieren damit vor allem eine Meibom-Drüsen-Dysfunktion, welche bei der SjD ebenfalls einen Teil der okulären Sicca-Symptomatik erklärt.22
Zur Verlängerung der Tränenfilmverweildauer und zur Reduktion des Tränenabflusses können sogenannte Punctum-Plugs in die Tränenkanälchen eingesetzt werden – temporär oder dauerhaft, je nach Verlauf. In schweren, therapierefraktären Fällen kann zudem autologes Serum in Tropfenform eingesetzt werden. Bei mittelschwerer bis schwerer Keratoconjunctivitis sicca kann auch eine topische Immunsuppression mit Cyclosporin-Augentropfen zum Einsatz kommen.
Ergänzend können nichtmedikamentöse Massnahmen wie regelmässige Lidrandpflege, das Tragen feuchtigkeitsstabilisierender Schutzbrillen («moisture chamber glasses») sowie eine Luftbefeuchtung in Innenräumen sinnvoll sein, um die Verdunstung des Tränenfilms zu reduzieren und die Beschwerden zu lindern.
Orale Sicca-Symptomatik
Die Therapie der oralen Trockenheit umfasst sowohl sekretersetzende als auch sekretstimulierende Massnahmen. Speichelersatzmittel wie Mundsprays, Lutschtabletten oder Gele mit befeuchtender Wirkung – etwa auf Basis von Carboxymethylcellulose oder Xylitol – können die subjektiven Beschwerden lindern und die Schleimhäute schützen. Ergänzend empfehlen sich das Kauen von zuckerfreiem Kaugummi oder der Verzehr saurer zuckerfreier Bonbons, um den Speichelfluss auf physiologischem Weg anzuregen.
SjD-Betroffene mit erhaltener Restfunktion der Speicheldrüsen kann die Gabe von Pilocarpin (5mg, drei- bis viermal täglich) oder Cevimelin (30mg, dreimal täglich – sofern verfügbar) zur Sekretanregung sinnvoll sein. Diese Substanzen sind jedoch mit potenziellen Nebenwirkungen wie vermehrtem Schwitzen, Flush oder Durchfällen verbunden und ihr Einsatz sollte daher individuell abgewogen werden.
Ein weiterer Schwerpunkt liegt auf der Prävention dentaler Komplikationen: Intensive Mundhygiene mit fluoridhaltiger Zahnpasta, regelmässige zahnärztliche Kontrollen sowie die Vermeidung kariogener Nahrungsmittel sind essenziell. Aufgrund der veränderten oralen Mikroflora und der reduzierten Abwehrlage ist zudem eine erhöhte Anfälligkeit für Candida-Infektionen zu beachten. Hier können lokal wirksame Antimykotika zum Einsatz kommen.
Systemische Therapie
Bei extraglandulären Manifestationen kann eine systemische, immunmodulierende resp. immunsuppressive Therapie indiziert sein. Die Auswahl der Medikamente richtet sich nach der Organbeteiligung, der Krankheitsaktivität und den individuellen Komorbiditäten.
Hydroxychloroquin stellt eine häufig gut verträgliche Therapie dar, insbesondere bei arthralgischen Beschwerden, Hautmanifestationen und zur Reduktion des Risikos des kongenitalen Leitungsblocks bei positiven Ro/SSA-Autoantikörpern. Regelmässige augenärztliche Kontrollen sind notwendig, da es sich bei der durch Hydroxychloroquin potenziell ausgelösten Retinopathie um eine seltene, aber schwerwiegende Komplikation handelt, die bei frühzeitiger Diagnosestellung in der Regel reversibel ist.
Bei aktiver systemischer Erkrankung kommen häufig Glukokortikoide zum Einsatz. Diese können initial in höherer Dosierung (z.B. Prednison 0,5–1mg/kgKG/d) verabreicht werden und sollten anschliessend zügig auf eine möglichst niedrige Erhaltungsdosis reduziert werden, um Langzeitnebenwirkungen zu vermeiden.
Konventionelle krankheitsmodifizierende Medikamente (DMARD) wie Methotrexat, Leflunomid, Azathioprin oder Mycophenolat-Mofetil können je nach den systemischen Organmanifestationen sinnvoll sein. Ihre Wirkung setzt oft verzögert ein, daher ist eine Kombination mit Glukokortikoiden zu Beginn üblich.
Bei schweren, therapierefraktären Verläufen oder bei bestimmten Risikokonstellationen (z.B. vaskulitische Polyneuropathie, ZNS-Beteiligung, lymphomverdächtige Befunde) kann der Einsatz von Biologika erwogen werden, wobei oft eine B-Zell-depletierende Therapie mit Rituximab oder eine BAFF-Inhibition mit Belimumab eingesetzt werden. Die Gabe erfolgt «off-label» und in der Regel in spezialisierten Zentren.
Ausblick
Die Forschung zur SjD hat in den letzten Jahren deutlich an Fahrt aufgenommen. Mehrere Phase-II-Studien konnten erstmals ihre primären Endpunkte erreichen – ein klares Signal für künftige therapeutische Fortschritte.
Beispielhaft ist der BAFF-Rezeptor-Antikörper Ianalumab, der in einer grossen, randomisierten, placebokontrollierten Phase-II-Studie eine signifikante Reduktion der Krankheitsaktivität (ESSDAI) und zusätzlich eine Verbesserung der Speichelflussrate erzielte.23 Auch der CD40-Antikörper Iscalimab, der gezielt in die Interaktion zwischen T- und B-Zellen eingreift, zeigte eine relevante klinische Wirksamkeit der Krankheitsaktivität (ESSDAI) in einer Proof-of-Concept-Studie.24
Weitere Wirkstoffe wie das RNase-Fc-Fusionsprotein RSLV-132 oder das «small molecule» Iguratimod werden aktuell in Phase-IIb-Studien untersucht. Ihre vielschichtigen Wirkmechanismen eröffnen neue therapeutische Möglichkeiten – von der gezielten Autoantikörperbindung bis zur Hemmung proinflammatorischer Signalwege.
Insgesamt lässt sich ein Trend hin zur stratifizierten Therapie erkennen, die immunpathologische Subgruppen differenzierter adressiert. Sollten sich diese innovativen Therapieansätze in grösseren Phase-III-Studien bestätigen, steht die Therapie der SjD vor einem Paradigmenwechsel.
Literatur:
1 Sjögren H: Zur Kenntnis der Keratoconjunctivitis sicca. Keratitis filiformis bei Hypofunktion der Tränendrüsen. Acta Ophthalmol 1933; 2: 1-151 2 Kivity S et al.: Infection and autoimmunity in Sjogren’s syndrome: a clinical study and comprehensive review. J Autoimmun 2014; 51: 17-22 3 Punnanitinont A, Kramer JM: Sex-specific differences in primary Sjögren’s disease. Front Dent Med 2023; 4: 1168645 4 Thompson N et al.: Exploring BAFF: its expression, receptors and contribution to the immunopathogenesis of Sjögren’s syndrome. Rheumatology 2016; 55: 1548-55 5 Napeñas JJ, Rouleau TS: Oral complications of Sjögren’s syndrome. Oral Maxillofac Surg Clin North Am 2014; 26: 55-62 6 Gau SY et al.: Higher risk for Sjögren’s syndrome in patients with fibromyalgia: a nationwide population-based cohort study. Front Immunol 2021; 12: 640618 7 Mæland E et al.: Understanding fatigue in Sjögren’s syndrome: outcome measures, biomarkers and possible interventions. Front Immunol 2021; 12: 703079 8 Melsens K et al.: Nailfold capillaroscopy in Sjögren’s syndrome: a systematic literature review and standardised interpretation. Clin Exp Rheumatol 2020; 38(Suppl 126): 150-7 9 Maślińska M et al.: Usefulness of rheumatoid factor as an immunological and prognostic marker in PSS patients. Clin Rheumatol 2019; 38: 1301-7 10 Yazisiz V et al.: Clinical and serological characteristics of seronegative primary Sjögren’s syndrome: a comparative study. Clin Rheumatol 2021; 40: 221-9 11 Lacombe V et al.: Unstimulated whole saliva flow for diagnosis of primary Sjögren’s syndrome: time to revisit the threshold? Arthritis Res Ther 2020; 22: 38 12 Finzel S et al.: Patient-based reliability of the Outcome Measures in Rheumatology (OMERACT) ultrasound scoring system for salivary gland assessment in patients with Sjögren’s syndrome. Rheumatology 2021; 60: 2169-76 13 Hočevar A et al.: Development of a new ultrasound scoring system to evaluate glandular inflammation in Sjögren’s syndrome: an OMERACT reliability exercise. Rheumatology 2022; 61: 3341-50 14 Ramsubeik K et al.: Diagnostic accuracy of salivary gland ultrasound in Sjogren’s syndrome: A systematic review and meta-analysis. Ther Adv Musculoskelet Dis 2020; 12: 1759720X20973560 15 Mossel E et al.: Ultrasonography of major salivary glands compared with parotid and labial gland biopsy and classification criteria in patients with clinically suspected primary Sjogren’s syndrome. Ann Rheum Dis 2017; 76: 1883-9 16 Kordossis T et al.: Prevalence of Sjögren’s-like syndrome in a cohort of HIV-1-positive patients: descriptive pathology and immunopathology. Br J Rheumatol 1998; 37: 691-5 17 Sène D et al.: Ectopic germinal center-like structures in minor salivary gland biopsy tissue predict lymphoma occurrence in patients with primary Sjögren’s syndrome. Arthritis Rheumatol 2018; 70: 1481-8 18 Quartuccio L et al.: Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun Rev. 2015; 14: 1019-22 19 Voulgarelis M et al.: Prognosis and outcome of non-Hodgkin lymphoma in primary Sjögren syndrome. Medicine 2012; 91: 1-9 20 Retamozo S et al.: Prognostic markers of lymphoma development in primary Sjögren syndrome. Lupus 2019; 28(8): 923-36 21 Ramos-Casals M et al.: EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann Rheum Dis 2020; 79: 3-18 22 Noh SR et al.: Meibomian gland atrophy with duration of Sjogren’s syndrome in adult females. Int Ophthalmol 2022; 42: 191-200 23 Bowman SJ et al.: Safety and efficacy of subcutaneous ianalumab (VAY736) in patients with primary Sjogren’s syndrome: a randomised, double-blind, placebo-controlled, phase 2b dose-finding trial. Lancet 2022; 399: 161-71 24 Fisher B et al.: Assessment of the anti-CD40 antibody iscalimab in patients with primary Sjögren’s syndrome: a multicentre, randomised, double-blind, placebo-controlled, proof-of-concept study. Lancet Rheumatol 2020; 2: e142-52
Das könnte Sie auch interessieren:
Ist Pelargonium sidoides eine Therapieoption bei Kindern mit Hand-Fuss-Mund-Krankheit?
Eine Studie hat den Pelargonium-sidoides-Wurzelextrakt EPs®7630 erstmals bei Kindern mit Hand-Fuss-Mund-Krankheit untersucht und eine signifikante Reduktion des Beschwerdegrads und der ...

Geriatrie-Assessment in der Praxis
Im Praxisalltag kann es für Hausärzt:innen mitunter schwierig sein, abzuschätzen, wie gut ihre älteren Patient:innen im Alltag noch funktionieren. Oft gibt erst die Fahreignungsprüfung ...

Stürze bei älteren Menschen – potenziell fatal, aber auch vermeidbar
Stürze bei älteren Menschen sind häufig und mit schwerwiegenden Folgen assoziiert, werden aber oft aufgrund von Schamgefühl und kognitiven Einschränkungen von den Betroffenen nicht ...