Néphrocalcinose, mutations, calculs rénaux, et le rôle de l’âge
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Le Pr Martin Konrad dirige le service de néphrologie pédiatrique de la Clinique universitaire pédiatrique de Münster. Lors du congrès annuel de la Société Suisse de Néphrologie (SSN), il a donné une conférence sur l’état des connaissances intitulée «Phenotypic Expression of Kidney Tubular Disorders. Age Matters!».
Keypoints
-
L’hypercalcémie infantile idiopathique se traduit par des taux de calcium très élevés dans le sérum et les urines. Les symptômes sont entre autres: néphrocalcinose, retard de croissance, hypotonie et déshydratation.
-
Ce trouble métabolique peut être causé par différentes anomalies génétiques qui perturbent directement ou indirectement la dégradation de la vitamine D3.
-
Les anomalies génétiques moins significatives sont plus fréquentes dans la population. Elles influencent l’équilibre phosphocalcique et contribuent ainsi à la formation de calculs rénaux.
-
Des mesures médicales simples, telles qu’une substitution en phosphate ou l’arrêt de la prophylaxie par la vitamine D3, semblent être efficaces.
Les deux enfants dont le rapport de cas a été présenté par le néphrologue pédiatrique M. Konrad dans son exposé n’avaient que trois et quatre mois lorsqu’on leur a diagnostiqué une hyperoxalurie primitive de type 1 (HP1) infantile. Un long parcours thérapeutique s’annonçait dès lors pour eux: en plus de la dialyse péritonéale quotidienne, ils devaient subir une hémodialyse tous les deux jours. L’objectif thérapeutique était d’éliminer l’oxalate de l’organisme. Une fois qu’ils avaient environ six mois, ils ont également reçu un double traitement d’interférence ARN. Il visait à inhiber la traduction d’un gène responsable de la production de glyoxylate, un substrat pour la formation d’oxalate.1
L’HP1 est une maladie rare et potentiellement mortelle pour les personnes atteintes. Cette maladie génétique est caractérisée par un déficit en une enzyme qui dégrade le glyoxylate. Sans cette enzyme, le glyoxylate se transforme en oxalate. Celui-ci se lie au calcium dans l’organisme, formant des cristaux insolubles qui se déposent dans les reins et se transforment en calculs rénaux. Une étude longitudinale a montré chez 192 participant·es atteint·es d’HP1, que la survie rénale était de 79% 10 ans après le diagnostic, et de 51% 20 ans après.2
Malgré le traitement par ARN: les deux enfants atteints d’HP1 présentaient des lésions rénales sévères causées par l’oxalate. L’un a subi une transplantation rénale à l’âge de deux ans et n’a plus besoin de dialyse depuis.1 Pour l’autre enfant, aucun organe compatible n’a encore été trouvé, a raconté M. Konrad.
La néphrocalcinose chez les enfants
Outre l’hyperoxalurie, comme celle dont souffrent les enfants du rapport de cas de M. Konrad, l’hypercalciurie représente également un risque de dépôts de calcium dans les reins (ou néphrocalcinose) et de calculs rénaux. «Les enfants sont plus susceptibles de développer une néphrocalcinose», a déclaré M. Konrad. En revanche, les calculs rénaux seraient plus fréquents chez les adultes. L’âge auquel une maladie rénale génétique se manifeste semble influencer l’expression phénotypique. «Age matters!», a-t-il souligné.
Le calcium est réabsorbé dans les reins: environ deux tiers par un transport passif dans le tube proximal, le reste dans le tube distal, en particulier dans la branche ascendante épaisse.3 La réabsorption dans le tube distal est régulée par la parathormone: elle vise à augmenter rapidement la concentration de calcium dans le plasma et favorise donc non seulement la réabsorption rénale, mais stimule également la synthèse de calcitriol (1,25-[OH]2-vitamine D3), la forme active de la vitamine D3. Le calcitriol favorise lui l’absorption intestinale du calcium et régule l’homéostasie osseuse (Fig.1).4
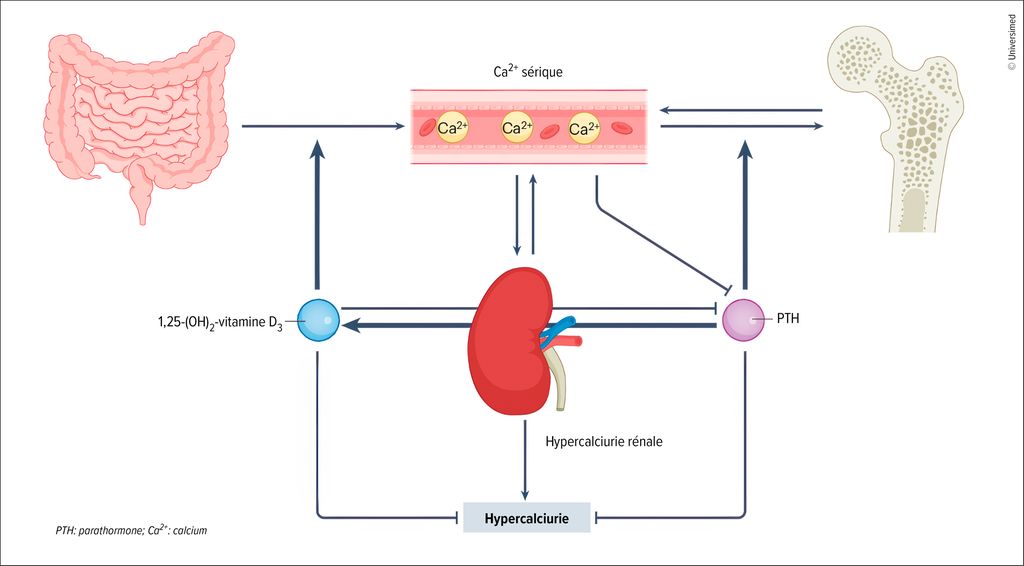
Fig.1: Physiopathologie de l’hypercalciurie. La réabsorption du calcium dans les reins se fait par un transport passif dans le tube proximal et un transport contrôlé par la parathormone dans le tube distal. La parathormone stimule également la synthèse de la forme active de la vitamine D3 calcitriol (1,25-[OH]2-vitamine D3). Ce dernier favorise lui l’absorption intestinale du calcium et régule l’homéostasie osseuse (modifiée selon Bargagli M et al. 2025)4
Les transporteurs impliqués dans la récupération du calcium peuvent présenter des mutations génétiques. Selon le transporteur, différentes sections du tubule sont affectées, par exemple la branche ascendante épaisse dans le syndrome de Bartter, et le tube contourné distal dans le syndrome de Gitelman. «Il n’est donc pas surprenant que ces mutations influencent le développement de la néphrocalcinose ou la formation de calculs rénaux», a déclaré M.Konrad. Il a gardé un souvenir particulier d’une patiente atteinte de néphrocalcinose. Ce cas a eu une influence déterminante sur ses propres recherches.
Quelle est la cause de l’hypercalcémie idiopathique?
M. Konrad a reçu pour la première fois la fillette en consultation il y a 20 ans, elle était alors âgée de six mois. En plus d’une néphrocalcinose, elle présentait une polyurie, une déshydratation, une hypotonie et un retard de croissance pour son âge. «Elle avait des taux de calcium significativement élevés dans le sérum et les urines, mais un faible taux de parathormone», a-t-il déclaré. On ne savait pas à l’époque quelle mutation était responsable de ce tableau clinique, ou même s’il s’agissait d’une maladie héréditaire. C’est ainsi que le diagnostic d’hypercalcémie infantile idiopathique (HII) a été posé chez la fillette. «Nous avons administré de l’acide pamidronique, arrêté l’administration de vitamine D3 et réduit l’apport en calcium par l’alimentation», a ajouté M.Konrad. Le taux de calcium a baissé et l’état de santé de l’enfant s’est amélioré.
Même si l’enfant allait mieux, l’HII ne laissait pas M. Konrad tranquille. «La maladie a été décrite pour la première fois dans les années 1950», a-t-il déclaré. «Durant cette période, la Grande-Bretagne a connu une augmentation endémique des cas d’HII.» Les autorités sanitaires britanniques recommandaient alors d’administrer aux enfants des doses élevées de vitamine D3, à savoir 4000UI/j. Dans d’autres pays, la recommandation était de 400UI/j. En raison de la fréquence de l’HII en Grande-Bretagne, on a supposé qu’une hypersensibilité à la vitamine D3 en était la cause.5
«L’HII nous intéressait, nous avons donc constitué une cohorte d’enfants concernés», a raconté M. Konrad. En se basant sur les résultats obtenus en Grande-Bretagne, il a supposé que la maladie était due à une mutation d’un gène impliqué dans le métabolisme de la vitamine D3. Lui et son équipe ont essayé de trouver le gène correspondant en utilisant une «candidate gene approach». La forme active de la vitamine D3 calcitriol est produite dans les reins à partir du calcidiol. Ce dernier est produit dans le foie à partir du cholécalciférol, qui est à son tour isomérisé par les rayons UVB dans la peau avant de passer dans la circulation sanguine.6
«Nous avons ainsi résolu deux énigmes»
M. Konrad et son équipe ont eu de la chance: ils ont effectivement trouvé une mutation dans le gène CYP24A1 chez six enfants atteints d’HII. Ce gène code pour l’enzyme 25-hydroxyvitamine D-24-hydroxylase. Cette dernière dégrade le calcitriol, mais perd sa fonction en cas de mutation.7 «Si on l’administre aux enfants porteurs de cette mutation, la vitamine D3 s’accumule et entraîne une hypercalcémie», a expliqué M. Konrad. De plus, les chercheur·ses ont également trouvé la mutation dans le gène CYP24A1 dans une deuxième cohorte de quatre enfants, qui avaient reçu une prophylaxie par bolus à forte dose de vitamine D3 et ont ensuite développé une hypercalcémie symptomatique. M. Konrad a conclu que la mutation n’était pas seulement un facteur de risque génétique d’hypercalcémie, mais que l’hypercalcémie pouvait aussi être provoquée par une prophylaxie par la vitamine D3.7 «Nous avons ainsi résolu deux énigmes: l’hypercalcémie infantile ‹désormais non idiopathique› et l’hypersensibilité à la vitamine D3», a-t-il déclaré.
Dans le cas de la mutation dans le gène CYP24A1, un seul allèle ou les deux allèles peuvent être affectés.8 Il a expliqué que le risque de développer une HII était élevé en cas de mutation biallélique «Si un seul allèle est touché, les symptômes semblent survenir un peu plus tard et sont alors plus susceptibles d’entraîner des calculs rénaux. Ici aussi, l’âge a une influence sur le phénotype.», a-t-il ajouté. Dans la cohorte de M.Konrad, il y avait cependant aussi des patient·es diagnostiqué·es d’HII, mais qui ne présentaient aucune mutation dans le gène CYP24A1, que ce soit hétérozygote ou biallélique. Cependant, le tableau clinique de ces patient·es était différent sur un point: il·elles présentaient tou·tes une hypophosphatémie.9
Le rôle du phosphate
Le phosphate est réabsorbé à 80–90% dans les reins. Les protéines de transport nécessaires à ce processus se trouvent principalement dans le tube proximal. Le phosphate récupéré agit sur le métabolisme de la vitamine D3: il inhibe la formation de la forme active de la vitamine D3 calcitriol et favorise sa dégradation. NaPi-2a est l’abréviation de cotransporteur sodium-phosphate de type 2a, qui est l’un des symports principaux de la réabsorption du phosphate. «Chez les personnes atteintes d’HII sans mutation dans le gène CYP24A1, nous avons trouvé une mutation dans le gène codant pour le NaPi-2a», a déclaré M. Konrad. Cela explique à la fois l’hypophosphatémie et l’hypercalcémie, poursuit M. Konrad. Pour le traitement, ils auraient d’abord seulement arrêté la supplémentation en vitamine D3. «Mais cela n’a pas eu beaucoup d’effet», a-t-il précisé. Ce n’est que l’administration de phosphate qui aurait finalement augmenté la concentration sérique tout en diminuant le calcium.9
Le défaut d’expression de NaPi 2a est causé par une anomalie génétique de SLC34A1. Comme pour CYP24A1, la mutation dans le gène SLC34A1 peut se produire sur un seul allèle ou sur les deux. Cela semble ici aussi avoir une influence sur le phénotype clinique. Une mutation biallélique implique un risque élevé d’HII. Si la mutation concerne un seul allèle, alors la maladie se manifeste plutôt par une néphrocalcinose et des calculs rénaux.10 «D’une manière très générale, on peut dire que les mutations à fort effet se manifestent sous forme de maladies. En revanche, les autres mutations entraînent plutôt des phénotypes subcliniques, voire sains, mais en contrepartie, elles sont aussi plus fréquentes dans la population que les mutations à fort effet», a conclu M. Konrad.
Source:
Congrès annuel de la Société Suisse de Néphrologie, du 4 au 5 décembre 2025, à Interlaken
Littérature:
1 Metry EL et al.: Successful kidney-alone transplantation in a patient with PH1 on combination RNA-interference therapy. Kidney Int 2023; 104: 203-4 2 Zhao F et al.: Predictors of incident ESRD among patients with primary hyperoxaluria presenting prior to kidney failure. Clin J Am Soc Nephrol 2015; 11: 119-26 3 Hanna RM et al.: Calcium transport in the kidney and disease processes. Front Endocrinol 2022; 12: 762130 4 Bargagli M et al.: Kidney stone disease: risk factors, pathophysiology and management. Nat Rev Nephrol 2025; 21: 794-808 5 Hypercalcaemia in infants and vitamin D. Br Med J 1956; 2: 149 6 Delrue C et al.: Vitamin D and vitamin D-binding protein in health and disease. Int J Mol Sci 2023; 24 :4642 7 Schlingmann KP et al.: Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med 2011; 365: 410-21 8 Molin A et al.: CYP24A1 mutations in a cohort of hypercalcemic patients: evidence for a recessive trait. J Clin Endocrinol Metab 2015; 100: E1343-52 9 Schlingmann KP et al.: Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol 2016; 27: 604-14 10 Lapointe JY et al.: NPT2a gene variation in calcium nephrolithiasis with renal phosphate leak. Kidney Int 2006; 69: 2261-7
Das könnte Sie auch interessieren:
Études actuelles sur le traitement de l’ILD
Le terme pneumopathie interstitielle (ILD) est un terme générique désignant un groupe important et hétérogène de maladies qui se manifestent principalement dans l’espace broncho- ...

Mise à jour 2026: vers la modification de la maladie
Un grand nombre de médicaments biologiques immunomodulateurs et de «small molecules» ont considérablement élargi l’éventail des options thérapeutiques dans les maladies inflammatoires ...
Schémas thérapeutiques de référence plus courts dans la tuberculose
La tuberculose (TB) constitue toujours un défi majeur à l’échelle mondiale. Malgré les progrès, le besoin de schémas thérapeutiques plus efficaces, plus courts et mieux tolérés reste ...