
Des progrès lents dans la prise en charge des pneumopathies interstitielles
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Les pneumopathies interstitielles constituent un groupe important et parfois mal compris d’affections du parenchyme pulmonaire, dont certaines présentent une évolution dramatique et peuvent entraîner la mort dans des délais relativement courts. Récemment, des traitements sont devenus disponibles pour certaines de ces maladies. Ils peuvent réduire considérablement la mortalité.
Selon le Prof. Killian Hurley de l’Hôpital Beaumont de Dublin, les pneumopathies interstitielles (PI, «interstitial lung disease» [ILD,]) peuvent être divisées, du point de vue étiologique, en deux grands groupes:
-
les pneumonies interstitielles idiopathiques, dont la cause, comme le nom l’indique, est inconnue,
-
ainsi que les PI qui peuvent être attribuées à des agents nocifs connus ou qui sont associées à d’autres maladies, telles que les maladies auto-immunes.1
Différents modèles d’évolution de la PI fibrosante
La PI la plus connue et la plus étudiée aujourd’hui est la fibrose pulmonaire idiopathique (FPI, «idiopathic pulmonary fibrosis» [IPF]). Elle survient généralement chez des personnes de plus de 60 ans et se caractérise par une fibrose progressive du poumon, une perte de la fonction pulmonaire et une mortalité élevée. La survie médiane se situe entre trois et cinq ans. Cependant, selon K. Hurley, au moins trois phénotypes différents peuvent être identifiés dans cette fourchette de survie: un avec un déclin rapide et continu de la fonction pulmonaire, un avec une évolution graduelle déterminée par des exacerbations ainsi qu’une forme un peu plus bénigne accompagnée d’une perte plus lente de la fonction pulmonaire.2 Cependant, cette évolution généralement défavorable n’est pas limitée à la FPI. Environ 40% des patients atteints d’autres PI développent une fibrose pulmonaire progressive au cours de la maladie, qui est similaire à une FPI sur le plan à la fois physiopathologique et clinique. Cela s’applique, par exemple, aux PI associées à des maladies rhumatismales.
Certaines PI sont classées comme principalement fibro-prolifératives, tandis que dans d’autres, l’inflammation figure initialement au premier plan, mais elles peuvent évoluer vers un phénotype fibro-prolifératif au fil du temps. Le facteur déterminant est la répétition des lésions épithéliales ou vasculaires avec une réparation incontrôlée. Il en résulte une immigration de fibroblastes, qui se différencient en myofibroblastes, ce qui entraîne une augmentation de la matrice extracellulaire. De quoi entraîner une rigidification du tissu et une perte de fonction des alvéoles. Lors d’une étape suivante, les macrophages et les lymphocytes sont recrutés dans le tissu endommagé et libèrent des facteurs profibrotiques qui contribuent à l’activation supplémentaire des fibroblastes. La rigidification croissante du poumon favorise ces processus, qui finissent par prendre leur autonomie et entraîner une cicatrisation croissante du poumon.3
Cliniquement, ce phénotype fibrosant de la PI se caractérise par une détérioration progressive des échanges gazeux, s’exprimant par une dyspnée croissante et une diminution des performances, allant jusqu’à une insuffisance respiratoire. Les différents pathomécanismes ont chacun des corrélations cliniques différentes. Les lésions vasculaires entraînent une hypertension pulmonaire à l’effort, la lésion des voies aérienne provoque une perte de volume pulmonaire et une augmentation du réflexe de toux, et les alvéoles pathologiquement modifiées sont responsables, entre autres, de la capacité de diffusion limitée. Une perte de 10% de la capacité vitale forcée est déjà un facteur prédictif important de mortalité, d’exacerbations et d’un certain nombre d’autres résultats défavorables. Le tableau histologique de la pneumopathie interstitielle commune (PIC, «usual interstitial pneumonia» [UIP]), avec aspect en rayon de miel («honey combing») à la tomographie à haute résolution (HRCT) représentant le tissu pulmonaire perdu, est typique de la FPI et d’autres PI à phénotype fibrosant.
De bonnes données en faveur de la cryobiopsie transbronchique
La HRCT, en combinaison avec le tableau clinique, joue un rôle important dans le diagnostic des PI. Néanmoins, diverses lignes directrices ont jusqu’à présent préconisé la biopsie comme diagnostic standard – sachant qu’il s’agit d’une biopsie chirurgicale accompagnée d’un risque de mortalité qui ne saurait être sous-estimé. La biopsie transbronchique à la pince, simple et relativement peu risquée, n’est pas adaptée au diagnostic des PI, étant donné qu’elle fournit trop rarement une histologie claire en raison de la petite taille des biopsies altérée par des artefacts de compression. Il est donc urgent de trouver des alternatives. L’une d’entre elles est la cryobiopsie transbronchique, une procédure bronchoscopique au cours de laquelle le tissu pulmonaire est gelé sur une cryosonde puis prélevé. De quoi permettre de retirer de plus gros morceaux de parenchyme pulmonaire et d’éviter les artefacts de compression. L’étude Cold Ice, publiée en 2019, a contribué à établir la cryobiopsie dans le diagnostic des PI.4 Le Prof. Antje Prasse, de la Faculté de médecine de Hanovre, fait référence à un article récent qui a examiné dans quelle mesure la cryobiopsie a amélioré la sécurité du diagnostic au sein des équipes pluridisciplinaires prenant en charge les patients atteints de PI. Le classement issu des lignes directrices actuelles a été utilisé: il classifie un diagnostic comme «confiant» s’il est sûr à 90%. Grâce à la cryobiopsie, un diagnostic peut être posé avec plus de 90% de certitude chez 53% des patients. La cryobiopsie s’avère donc supérieure à la fois au tableau clinique plus HRCT et au lavage bronchoalvéolaire. Des diagnostics provisoires avec un niveau de confiance élevé peuvent même être établis chez 81% des patients.5 La cryobiopsie s’est également révélée être un atout important lors des réunions de l’équipe multidisciplinaire.
Un autre article récent a cherché à savoir si un aspect de PIC détecté dans la cryobiopsie prédit la mortalité aussi bien qu’un aspect de PIC dans la biopsie chirurgicale et a confirmé la valeur pronostique élevée de la cryobiopsie (Fig. 1).6 Selon A.Prasse, toutes ces données ont été intégrées dans la ligne directrice récemment publiée sur la cryobiopsie transbronchique. Celle-ci inclut également une méta-analyse sur la sécurité de la méthode, qui a constaté un risque de pneumothorax de 9,5% et d’hémorragies sévères de 1,1%.7 Pour A.Prasse, il en résulte un rapport bénéfice-risque très favorable, qui plaide en faveur de l’inclusion de la cryobiopsie dans le diagnostic des PI.
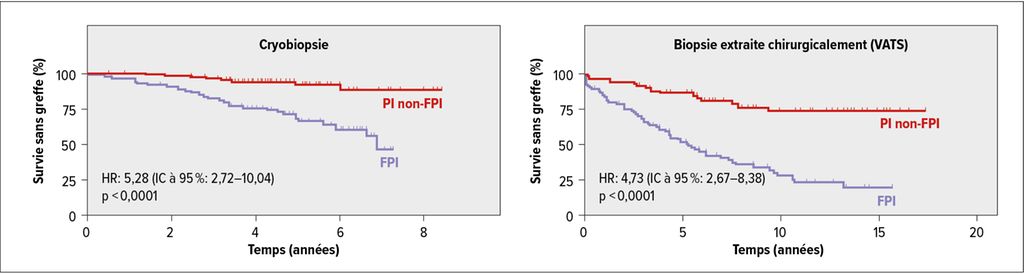
Fig. 1: Prédiction de la mortalité en fonction des caractéristiques de la PIC: cryobiopsie contre biopsie chirurgicale (modifié d’après Tomassetti et al.)6
Une autre étude récente porte sur le diagnostic de la PIC à l’aide de marqueurs biochimiques. Grâce à un classificateur génomique basé sur 160 gènes, une PIC peut être diagnostiquée à partir de la biopsie avec une haute spécificité. Cependant, la sensibilité n’est que de 63% – ce qui, selon A. Prasse, n’est pas ce que l’on attendrait pour l’exclusion d’une PIC. Cependant, la méthode pourrait potentiellement fournir une certitude supplémentaire dans le contexte du processus complet de diagnostic.
Le traitement antifibrotique réduit la mortalité y compris à long terme
En ce qui concerne le traitement de la PI, aucune bonne nouvelle n’a pu être annoncée ces derniers mois. Deux grands essais ont été interrompus en raison de problèmes d’innocuité. Le programme de phase III ISABELA avec le ziritaxestat a été interrompu prématurément étant donné que, de l’avis du comité de surveillance des données, les bénéfices du traitement ne justifiaient pas les risques. Dès la phase II, les études sur STX-100, un anticorps contre le récepteur de l’intégrine αVβ6, ont été interrompues pour cause de toxicité.
En revanche, il existe des données réelles encourageantes sur les traitements antifibrotiques de la FPI déjà approuvés qui démontrent également une réduction de la mortalité sur une plus longue période.9 A.Prasse souligne que l’efficacité du nintédanib et de la pirfénidone a été démontrée non seulement en cas de FPI, mais aussi dans d’autres formes de PI. Le nintédanib est donc déjà approuvé pour certaines autres formes de PI, comme les PI associées à la sclérose systémique. A. Prasse souligne que, compte tenu de l’autorisation désormais très large du nintédanib, il ne faut pas renoncer à un diagnostic exact et à une prise de décision clinique correcte.
Selon A. Prasse, ces données ont permis de définir une approche de traitement qui distingue trois groupes de PI: alors que les PI purement inflammatoires nécessitent un traitement immunosuppresseur, les patients présentant un phénotype fibrotique («IPF-like») bénéficient exclusivement de thérapies antifibrotiques. Un troisième groupe est celui des PI telles que celles associées à la sclérose systémique («SSc-like»), qui doivent être traitées à la fois par immunosuppression et dans une démarche antifibrotique.
Pas d’épidémie de PI à la suite du Covid-19
Les pneumologues et les infectiologues se sont beaucoup inquiétés de comptes-rendus de patients qui ont développé une PI au cours d’une infection à Covid-19, dans le cadre de laquelle la HRCT a initialement révélé des opacités en verre dépoli qui se sont transformées, avec le temps, en aspect en rayon de miel typique d’une PIC. La crainte dans cette situation était de faire face à une vague de pneumopathies fibrosantes, selon A. Prasse. Mais heureusement, l’expérience montre désormais que ces lésions régressent avec le temps chez des individus auparavant sains, ce qui implique toutefois une longue période de convalescence. Les lésions fibrotiques sont décelables pendant des mois après la maladie aiguë. Les facteurs de risque sont l’âge et la gravité de la maladie – et surtout une insuffisance respiratoire aiguë. Cependant, une PI préexistante avant le Covid-19 est associée à une augmentation de la mortalité par Covid, les patients plus âgés étant particulièrement touchés.10
Source:
Satellites virtuels de l’ERS, 2 mars 2021
Littérature:
1 Kalchiem-Dekel O et al.: J Clin Med 2018; 7: 476 2 Maher TM: Eur Respir Rev 2013; 22: 148-52 3 McLean-Tooke A et al.: Clin Transl Immunology 2019; 8: e1086 4 Troy LK et al. Lancet Respir Med 2020; 8: 171-81 5 Hetzel J et al.: Eur Respir J 2020; 56: 1901520 6 Tomassetti S et al.: Lancet Respir Med 2020; 8: 786-94 7 Maldonado F et al.: Chest 2020; 157: 1030-42 8 Rixheldi L et al.: Am J Respir Crit Care Med 2021; 203: 211-20 9 Behr J et al.: Eur Respir J 2020; 56: 1902279 10 Drake TM et al.: Am J Respir Crit Care Med 2020; 202: 1656-65
Das könnte Sie auch interessieren:

L’asthme et le rythme circadien
Le rythme circadien ne joue pas seulement un rôle important dans le sommeil, il a également une influence considérable sur les crises d’asthme et la fonction pulmonaire. Le ciblage ...

Technologies avancées et existantes dans la gestion du diabète
Même si le terme «traitement» est compris dans le nom de la conférence, la 18th International Conference on Advanced Technologies & Treatments for Diabetes (ATTD) constitue le rendez- ...
Les défis du diabète de type 1
Dans le cas du diabète de type 1, surtout s’il ne se déclare qu’à l’âge adulte, la pose du diagnostic peut déjà constituer un défi. Le risque d’hypoglycémie, qu’il faut minimiser, et le ...