
Kardiale Amyloidose: frühzeitig erkennen & gezielt abklären
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die kardiale Amyloidose ist keine seltene Erkrankung, sondern eine häufig übersehene Ursache von Herzinsuffizienz und Myokardverdickung. Angesichts der heute verfügbaren krankheitsmodifizierenden Therapien ist eine frühzeitige Diagnosestellung von entscheidender prognostischer Bedeutung. Ein strukturierter diagnostischer Ansatz ermöglicht die rasche Differenzierung zwischen AL- und ATTR-Amyloidose und trägt dazu bei, betroffene Patientinnen und Patienten zeitnah einer adäquaten Therapie zuzuführen.
Keypoints
-
Eine kardiale Amyloidose muss bei Herzinsuffizienz mit erhöhter Wanddicke, insbesondere im höheren Lebensalter und bei Vorliegen kardialer oder extrakardialer „red flags“, aktiv und frühzeitig in Betracht gezogen werden.
-
Zunächst muss eine AL-Amyloidose ausgeschlossen werden, da die kardiale AL-Amyloidose unbehandelt rasch progredient verläuft.
-
Die Bestimmung freier Leichtketten im Serum einschließlich der κ/λ-Ratio sowie eine Immunfixation in Serum und Urin sind dafür essenziell.
-
Eine Perugini-Grad-2/3-Anreicherung ist nur dann nichtinvasiv diagnostisch für eine ATTR-Amyloidose, wenn keine monoklonale Gammopathie vorliegt.
-
Bei Nachweis einer Monoklonalität oder nicht eindeutigen bildgebenden Befunden ist die histologische Sicherung mit anschließender Typisierung der entscheidende diagnostische Schritt.
Die Amyloidose ist eine systemische Speichererkrankung, bei der fehlgefaltete Proteine in Form unlöslicher Amyloidfibrillen in verschiedenen Organen abgelagert werden. Ist das Myokard betroffen, entsteht eine infiltrative restriktive Kardiomyopathie mit progredienter diastolischer und im weiteren Verlauf auch systolischer Dysfunktion, Arrhythmien und Herzinsuffizienz. Klinisch stehen heute zwei Entitäten im Vordergrund: die Leichtketten-Amyloidose (AL) und die Transthyretin-Amyloidose (ATTR), Letztere mit einer hereditären Form (ATTRv) und einer Wildtypform (ATTRwt). Gemeinsam sind AL- und ATTR-Amyloidose für den ganz überwiegenden Teil der klinisch relevanten kardialen Amyloidosen verantwortlich (>98%).1
Transthyretin ist ein überwiegend in der Leber synthetisiertes Transportprotein für Thyroxin und das Retinol-bindende Protein. Physiologisch liegt es als stabiles Tetramer vor. Im Rahmen der ATTR-Amyloidose kommt es entweder infolge pathogener TTR-Mutationen bei der hereditären Form (ATTRv) oder infolge einer meist altersbedingten Tetramerinstabilität bei der Wildtypform (ATTRwt) zur Dissoziation in Monomere, die fehlfalten und sich als Amyloidfibrillen im Myokard ablagern. Die AL-Amyloidose entsteht hingegen auf dem Boden einer klonalen Plasmazellerkrankung mit Produktion amyloidogener monoklonaler Immunglobulin-Leichtketten. Die sichere Unterscheidung beider Formen ist essenziell, da sie sich sowohl in ihrer Pathophysiologie als auch in Prognose und Therapie grundlegend unterscheiden.
Kardiale Amyloidose gilt nicht mehr als bloße Rarität, sondern als häufig übersehene Ursache einer Herzinsuffizienz mit erhöhter Wanddicke. In einer systematischen Übersichtsarbeit und Metaanalyse von Screeningstudien zeigte sich bei Patientinnen und Patienten mit HFpEF eine gepoolte Prävalenz der kardialen Amyloidose von 15,1%. In Einzelstudien wurde bei Patienten >60. Lebensjahr mit linksventrikulärer Hypertrophie eine ATTR-Kardiomyopathie in bis zu 13% der Fälle nachgewiesen. Auch bei schwerer Aortenstenose ist die Prävalenz relevant: in TAVI-nahen Kollektiven wurde eine begleitende kardiale Amyloidose in etwa 10–15% der Fälle beschrieben. Diese Zahlen unterstreichen, dass die Erkrankung im klinischen Alltag aktiv gesucht werden muss.
Eine frühere Diagnosestellung ist prognostisch entscheidend. Insbesondere die kardiale AL-Amyloidose verläuft unbehandelt hochaggressiv. Besteht zum Zeitpunkt der Diagnose eine Herzinsuffizienz, liegt das mediane Überleben ohne Therapie bei etwa sechs Monaten. Umgekehrt zeigen aktuelle Daten zur ATTR-Kardiomyopathie, dass Patientinnen und Patienten heute häufiger in früheren Krankheitsstadien diagnostiziert werden und dies mit einer deutlich niedrigeren Mortalität einhergeht.3
Wann sollte an eine kardiale Amyloidose gedacht werden?
Der klinische Verdacht entsteht meist aus der Kombination kardialer und extrakardialer „red flags“ (Tab.1).4 Besonders verdächtig ist eine anderweitig nicht erklärte linksventrikuläre Wandverdickung bei älteren Patientinnen und Patienten mit HFpEF oder Aortenstenose, insbesondere bei zusätzlichen „red flags“. Echokardiografisch sind eine diastolische Dysfunktion, ein klassisches longitudinales Strainmuster mit relativem apikalem Sparing, rechtsventrikuläre Mitbeteiligung, verdickte Klappen oder ein kleiner Perikarderguss suggestiv. Auf EKG-Ebene sprechen ein Pseudoinfarktmuster, eine im Verhältnis zur Wanddicke diskrepant niedrige QRS-Spannung sowie frühe Überleitungsstörungen für eine Amyloidose. Als extrakardiale Symptome sind insbesondere folgende zu nennen: bilaterales Karpaltunnelsyndrom, Lumbalspinalkanalstenose, Bizepssehnenruptur, autonome oder periphere Neuropathie, Proteinurie, Makroglossie, positive Familienanamnese.4
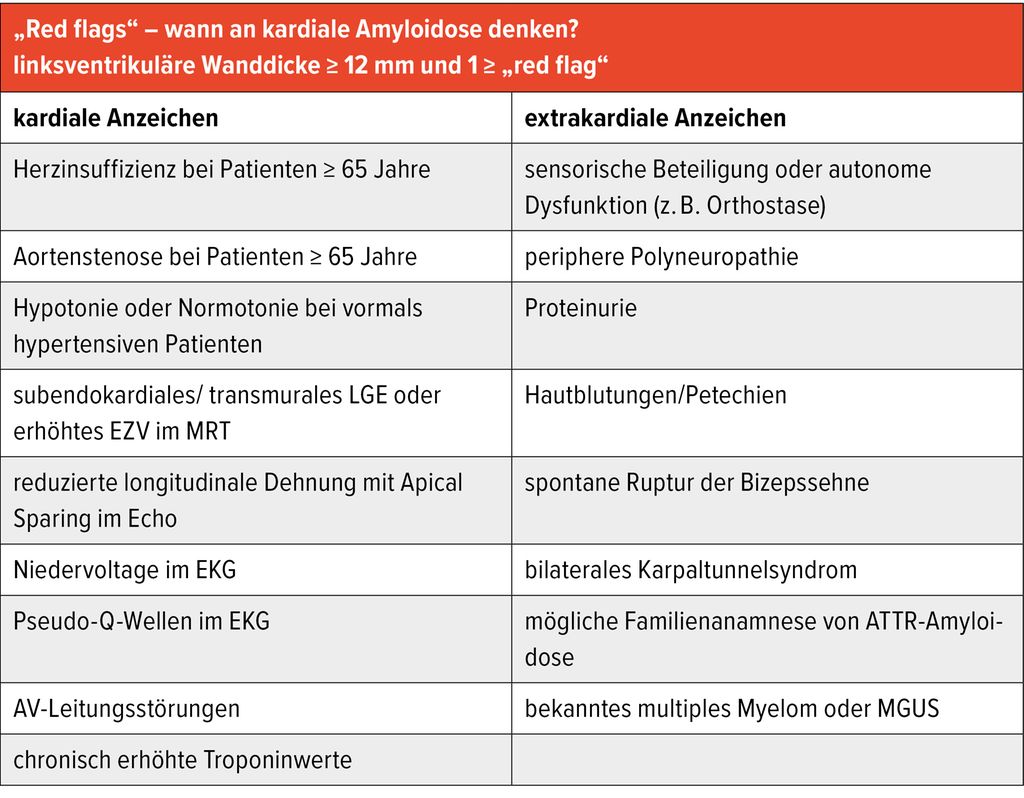
Tab. 1: Kardiale und extrakardiale „red flags“ (nach Arbelo E et al. 2023)4
Erster diagnostischer Schritt: AL-Amyloidose ausschließen
Bei Verdacht auf eine kardiale Amyloidose hat der rasche Ausschluss einer AL-Amyloidose oberste Priorität. Erforderlich sind die Bestimmung der freien Leichtketten im Serum einschließlich κ/λ-Ratio sowie die Immunfixation im Serum und Urin. Nur die Kombination dieser drei Untersuchungen erlaubt einen sensitiven Nachweis bzw. Ausschluss einer zugrunde liegenden monoklonalen Gammopathie. Bei eingeschränkter Nierenfunktion ist die freie Leichtkettenratio mit Vorsicht zu interpretieren.5 Bei grenzwertigen oder widersprüchlichen Befunden empfiehlt sich ein frühzeitiges hämatoonkologisches Konsil.
Knochenszintigrafie: zentraler Baustein der ATTR-Diagnostik
Parallel oder unmittelbar anschließend erfolgt die Knochenszintigrafie mit 99mTc-markierten Tracern (z.B. Technetium-99m-3,3-Diphosphono-1,2-Propandicarbonsäure [99mTc-DPD], Technetium-99m-Hydroxy-Methylen-Diphosphonat [HMDP] oder Technetium-99m-Pyrophosphat [PYP]), abhängig von der regionalen Verfügbarkeit. Die visuelle Bewertung erfolgt semiquantitativ anhand des Perugini-Scores: Grad 0 entspricht einer fehlenden myokardialen Anreicherung, Grad 1 einer geringeren Aufnahme als im Knochen, Grad 2 einer der Rippenanreicherung vergleichbaren Aufnahme und Grad 3 einer stärkeren myokardialen als knöchernen Anreicherung mit reduzierter oder fehlender Rippenaufnahme. Entscheidend ist, dass die Untersuchung planar und tomografisch interpretiert wird. Die SPECT- bzw. SPECT/CT-Komponente wird ausdrücklich empfohlen, um eine echte myokardiale Traceraufnahme sicher von Blutpool- oder Weichteilartefakten abzugrenzen.6 Eine Perugini-Grad-2- oder Perugini-Grad-3-Anreicherung ist stark hinweisend auf eine ATTR-Kardiomyopathie, jedoch nur dann nichtinvasiv diagnostisch, wenn keine monoklonale Gammopathie nachweisbar ist. Denn auch eine AL-Amyloidose kann prinzipiell mit unterschiedlichen „Uptake“-Graden einhergehen. Genau deshalb darf eine positive Szintigrafie niemals ohne parallele hämatologische Abklärung interpretiert werden.
Vier klinisch relevante Befundkonstellationen
Die häufigsten klinisch relevanten Befundkonstellationen sind:
-
Die Szintigrafie und Monoklonalitätsdiagnostik sind negativ. Eine kardiale Amyloidose ist dann unwahrscheinlich. Bei anhaltend hohem klinischem Verdacht – etwa bei typischem Phänotyp oder ausgeprägten extrakardialen Hinweisen – sollte dennoch eine weiterführende Abklärung mittels Herz-Magnetresonanztomografie (CMRT) und gegebenenfalls Biopsie erwogen werden, da einzelne ATTR-Varianten oder seltene Amyloidoseformen szintigrafisch negativ sein können.
-
Die Szintigrafie ist positiv, die Monoklonalitätsdiagnostik jedoch negativ. Bei Perugini-Grad 2 oder Grad 3 kann die Diagnose einer ATTR-Kardiomyopathie nichtinvasiv gestellt werden (Abb. 2). Anschließend sollte unabhängig vom Alter eine genetische Testung erfolgen, um zwischen ATTRv und ATTRwt zu unterscheiden. Bei Perugini-Grad 1 ist eine nichtinvasive Diagnose dagegen nicht möglich. Dies kann sowohl bei einer frühen ATTR-Kardiomyopathie als auch bei einer AL-Amyloidose vorkommen und erfordert daher eine weiterführende Abklärung, gegebenenfalls mit histologischer Sicherung und Typisierung.
-
Die Szintigrafie ist negativ und Monoklonalitätsdiagnostik positiv. In dieser Konstellation muss eine AL-Amyloidose rasch ausgeschlossen oder bestätigt werden. Die Herz-MRT kann dabei helfen, eine kardiale Mitbeteiligung zu stützen, bleiben die Befunde jedoch suggestiv oder unklar, ist eine Biopsie mit Amyloidtypisierung erforderlich. Da die Zeit bis zur Diagnosesicherung bei AL prognostisch kritisch ist, sollte die histologische Sicherung nicht verzögert werden.
-
Die Szintigrafie und Monoklonalitätsdiagnostik sind positiv. Dies ist die diagnostisch komplexeste Konstellation. Möglich sind eine ATTR-Kardiomyopathie mit begleitender MGUS, eine AL-Amyloidose oder sogar eine Koexistenz beider Entitäten. In diesem Fall ist eine histologische Diagnosesicherung mit sicherer Typisierung obligat, meist mittels Endomyokardbiopsie in einem erfahrenen Zentrum. Genau hier liegt einer der häufigsten Fehler in nicht spezialisierten Settings: Eine positive Szintigrafie bei gleichzeitig pathologischer Monoklonalitätsdiagnostik darf nicht automatisch als ATTR interpretiert werden (Abb. 1)

Abb. 1: Diagnostischer Algorithmus der kardialen Amyloidose (nach Garcia-Pavia P et al. 2021)1
Abb. 2: Nichtinvasive Diagnostik der Transthyretin-Amyloidose (adaptiert nach Arbelo E et al. 2023, Fontana M et al. 2025, Garcia-Pavia P et al. 2021 & Munder M et al. UniversitätsKlinikum Heidelberg)1
Die Herz-MRT hat in der modernen Amyloidosediagnostik einen hohen Stellenwert, insbesondere bei unklaren oder widersprüchlichen Befundkonstellationen. Charakteristisch sind ein diffuses subendokardiales oder transmurales „late gadolinium enhancement“ (LGE), eine abnorme Gadoliniumkinetik mit veränderter Myokardnullung sowie erhöhte native T1-Werte und ein vergrößertes extrazelluläres Volumen. In einer multizentrischen Studie zeigte das LGE-CMRT gegenüber der Endomyokardbiopsie eine Sensitivität von 95% und eine Spezifität von 98% für das Vorliegen einer kardialen Amyloidose.7 Gleichwohl bleibt die Biopsie Goldstandard, wenn die Subtypisierung unklar ist oder zwischen AL und ATTR sicher unterschieden werden muss.
Fazit
Die Diagnostik der kardialen Amyloidose folgt heute einem klar strukturierten, überwiegend nichtinvasiven Stufenkonzept. Entscheidend ist, die Erkrankung frühzeitig überhaupt zu vermuten, AL-Amyloidose sofort laborchemisch abzuklären und die Knochenzintigrafie nur zusammen mit SPECT und Monoklonalitätsdiagnostik zu interpretieren. Eine nichtinvasive ATTR-Diagnose ist nur bei kompatibler Bildgebung, Perugini-Grad 2/3 und fehlendem Nachweis einer monoklonalen Gammopathie zulässig. Gerade weil heute wirksame krankheitsmodifizierende Therapien zur Verfügung stehen, ist eine konsequente und präzise Diagnostik prognostisch von zentraler Bedeutung.
Literatur:
1 Garcia-Pavia P et al.: Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2021; 42(16): 1554-68 2 Aimo A et al.: Redefining the epidemiology of cardiac amyloidosis. A systematic review and meta-analysis of screening studies. Eur J Heart Fail 2022; 24(12): 2342-51 3 Ioannou A et al.: Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation 2022; 146(22): 1657-70 4 Arbelo E et al.: 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J 2023; 44(37): 3503-626 5 Milani P et al.: Diagnostic performance of the new free light chain ratio in systemic amyloidosis. J Am Coll Cardiol CardioOnc 2025; 7(6): 751-9 6 Khor et al.: 99mTc bone-avid tracer cardiac scintigraphy: role in noninvasive diagnosis of transthyretin cardiac amyloidosis. Radiology 2023; 306(2): e221082 7 Chatzantonis G et al.: Diagnostic value of cardiovascular magnetic resonance in comparison to endomyocardial biopsy in cardiac amyloidosis: a multi-centre study. Clin Res Cardiol 2021; 110(4): 555-68
Das könnte Sie auch interessieren:

ESC gibt umfassende Empfehlung für den Sport
Seit wenigen Tagen ist die erste Leitlinie der ESC zu den Themen Sportkardiologie und Training für Patienten mit kardiovaskulären Erkrankungen verfügbar. Sie empfiehlt Training für ...
ESC-Guideline zur Behandlung von Herzvitien bei Erwachsenen
Kinder, die mit kongenitalen Herzvitien geboren werden, erreichen mittlerweile zu mehr 90% das Erwachsenenalter. Mit dem Update ihrer Leitlinie zum Management kongenitaler Vitien bei ...

Inclisiran bei Patienten mit Statinintoleranz wirksam und sicher
Eine Analyse statinintoleranter Patienten aus dem Phase III Studienprogramm ORION zeigt, dass Inclisiran die LDL-Cholesterinspiegel kardiovaskulärer Hochrisikopatienten, die kein Statin ...