
Myelodysplastische Syndrome: neuer Name, neue Klassifikation
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Myelodysplastische Syndrome gehören zu den häufigsten Neoplasien. Die Bezeichnung ist jedoch nicht mehr ganz korrekt, obwohl sie noch verwendet wird. Welche weiteren Neuerungen es gibt und wie sich die Behandlung von Niedrig- und Hochrisiko-MDS unterscheidet, lesen Sie hier.
Das MDS ist definiert als heterogene Gruppe klonaler hämatopoetischer Stammzellerkrankungen, die gekennzeichnet sind durch eine dysplastische und insuffiziente Hämatopoese im Knochenmark (KM). Die Folgen sind periphere Zytopenien und ein Risiko für Progression in eine AML. Oftmals ist die Erstmanifestation die Anämie. MDS gehören zu den häufigsten hämatologischen Neoplasien. Die Inzidenz in der Gesamtbevölkerung in Deutschland liegt bei 4–5 pro 100000 Einwohner:innen jährlich. In der Altersgruppe der über 75-Jährigen steigt sie auf über 30 pro 100000 Einwohner:innen an. Das mediane Alter bei Erstdiagnose liegt bei etwa 75 Jahren. Männer sind häufiger betroffen als Frauen.1, 2
Änderungen beim Name und bei der Klassifikation
Entsprechend der fünften Edition der WHO-Klassifikation wird der Begriff „ myelodysplastische Syndrome“ durch „myelodysplastische Neoplasie“ ersetzt, um dem neoplastischen Charakter mehr Geltung zu geben.1, 2 Die Abkürzung „MDS“ wird aber inkonsequenterweise beibehalten. Um dem immer größeren Einfluss molekularer und genetischer Aberrationen auf die Prognose und Therapie des MDS besser gerecht zu werden, wurde das MDS in die beiden Gruppen der „genetisch definierten MDS“ und der „morphologisch definierten MDS“ unterteilt. Die Gruppe der „genetisch definierten MDS“besteht aus drei Subgruppen:
-
MDS mit niedriger Blastenzahl (unter 5 % im KM und unter 2 % im Blut) und isoliertem 5q-Syndrom,
-
MDS mit niedriger Blastenzahl und SF3B1-Mutation (vormals ringsideroblastisches MDS),
-
prognostisch ungünstige MDS mit bi-allelischer TP53-Mutation.
Die morphologisch definierte Gruppe der MDS enthält weitere fünf Subgruppen: erstens die Subgruppe mit niedriger Blastenzahl mit einem Blastenanteil von weniger als 5%. Zweitens MDS mit erhöhtem Blastenanteil von 5–9% im KM und 2–4% im Blut („increased blasts 1“ [IB1]); sowie drittens mit Blastenanteilen von 10–19% im KM und 5–19% im Blut („increased blasts 2“ [IB2]). Die vierte Gruppe stellt das hypoplastische MDS mit Blastenanteil unter 5% sowie altersadaptierter Zellularität von weniger als 25% dar. Als fünfte Subgruppe ist das MDS mit Fibrose mit einem Blastenanteil von mehr als 5% und einem Fibrosegrad von 2–3 definiert.1–2
In der neuen WHO-Klassifkation wurde bei den MDS/MPN(myeloproliferative Neoplasien)-Überlappungssydromen die Gruppe MDS/MPN-RS-T (mit Ringsideroblasten und Thrombozytose) nach WHO 2016 als „MDS/MPN mit SF3B1-Mutation und Thrombozytose“ umbenannt. Bei der chronischen myelomonozytären Leukämie (CMML) wurde die Einteilung CMML-0 wegen fehlender prognostischer Bedeutung wieder abgeschafft. Die Kategorie CMML-1 ist mit weniger als 10% Blasten im KM definiert und die Kategorie CMML-2 mit mehr als 10% Blasten.1–2
Klonale Hämatopoese als Frühform des MDS
Als Frühformen des MDS werden Subgruppen bezeichnet, die entweder eine Zytopenie oder eine zytogenetische oder molekulare Veränderung aufweisen, ohne jedoch die MDS-Kriterien in vollem Umfang zu erfüllen. Folgende vier Frühformen sind von Bedeutung:
-
Die ICUS („idiopathic cytopenia of undetermined siginificance“) geht einher mit einer Zytopenie, ohne molekulare oder zytogenetische Aberrationen aufzuweisen.
-
Die IDUS („idiopathic dysplasia undetermined significance“) weist lediglich dysplastische Veränderungen im KM ohne Zytopenien oder molekulare oder zytogenetische Aberrationen auf.
-
Die CHIP („Clonal hematopoiesis of indeterminate potential“) geht einher mit dem Nachweis von molekularen Mutationen, ohne Zytopenien aufzuweisen. Häufig lassen sich die Mutationen DNMT3A, TET2, ASXL1 nachweisen.3–4 CHIP stellt zwar definitionsgemäß eine gutartige Veränderung dar, hat aber ein erhöhtes Risiko, in ein MDS überzugehen. Wichtig ist, dass der Nachweis einer CHIP auch mit gesteigerten kardio-vaskulären Komplikationen assoziiert ist. Die Mortalität ist somit gegenüber der Normalpopulation deutlich erhöht.5,6
-
Die CCUS („Clonal cytopenia of undetermined significance“) weist neben molekularen Aberrationen auch eine Zytopenie auf.3–6
Neuer IPSS-M-Prognosescore
Der Prognosescore des neuen IPSS-M („molecular international prognostic scoring system“) umfasst die klinischen Faktoren medullärer Blastenanteil, Thrombozytenzahl, Hämoglobin sowie die zytogenetische Risikokategorie entsprechend dem IPSS-R(revised). Zusätzlich schließt er 17 molekulargenetische Variablen in 16 Genen sowie weitere 15 mutierte Gene von Bedeutung ein, sodass der Mutationsstatus von insgesamt 31 Genen in die Auswertung miteinfließt. Mit der Identifikation von sechs Risikogruppen stellt der neue Prognosescore eine signifikante Verbesserung der prognostischen Präzision in Bezug auf das Gesamtüberleben, das leukämiefreie Überleben und das Risiko für Transformation zu einer AML(akute myeloische Leukämie).
Der IPSS-M-Score lässt sich webbasiert errechnen: https://mds-risk-model.com .7,8
Therapie des Niedrigrisiko-MDS
Das primäre Therapieziel beim Niedrigrisiko-MDS ist die Verbesserung der Lebensqualität der Patient:innen. Entsprechend werden nur symptomatische, oft transfusionspflichtige MDS-Patient:innen behandelt. Polytransfundierte Patient:innen sind längerfristig durch die begleitende sekundäre Hämochromatose (Kardiomyopathie) gefährdet. Bei Patient:innen, die mindestens 20 Erythrozytenkonzentrate erhalten bzw. einen Serumferritin-Spiegel von >1000ng/ml haben, sollte eine Therapie mit Eisenchelatoren (Deferasirox, Desferoxamin) erwogen werden. Insbesondere gilt dies dann, wenn eine allogene Stammzelltransplantation geplant ist, die bei Eisenüberladung mit einer erhöhten Mortalität assoziiert ist. Hochdosierte Erythropoetin-Applikationen (40000–80000IE/Woche) sind bei niedrigem endogenem Erythropoetin-Spiegel im Serum unter 100U/L besonders aussichtreich, die Erythrozyten-Transfusionsfrequenz zu senken.9–13
Lenalidomid ist bei transfusionspflichtigen MDS mit del(5q) zugelassen. Patient:innen mit hypoplastischem MDS können mit einer immunsuppressiven Therapie behandelt werden, wenn sie für eine allogene Stammzelltransplantation ungeeignet sind. Etwa 30% der Patient:innen sprechen auf Antithymozytenglobulin (ATG) und Cyclosporin A an.14
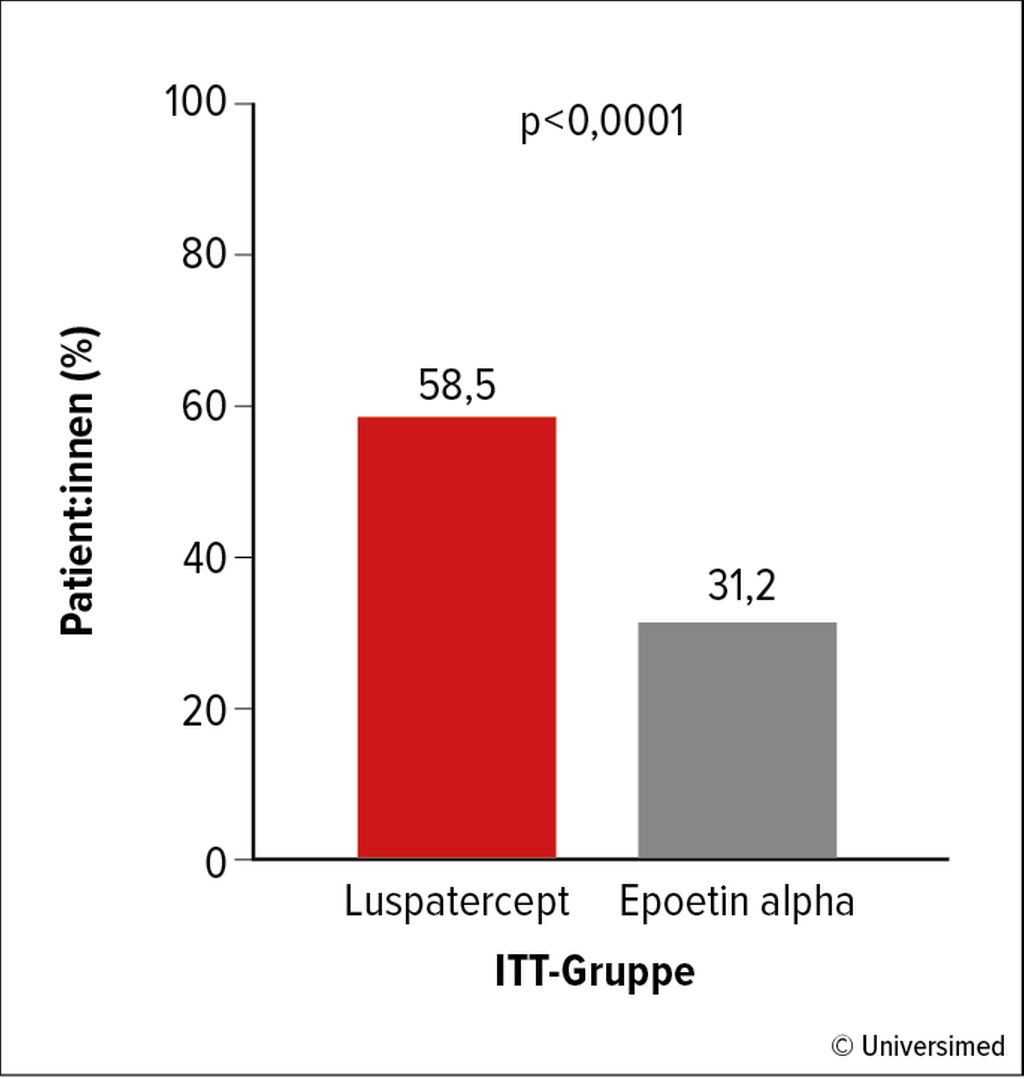
Abb. 1: Prozent der Patient:innen mit Transfusionsunabhängigkeit (TI; >12 Wochen) und Hb-Anstieg (>1,5g/dl) unter Luspatercept vs. Epoetin alpha bei ESA-naivem, transfusionsabhängigem Niedrigrisiko-MDS in der COMMANDS-Studie. Modifiziert nach Garcia-Manero G et al.16
Luspatercept, ein Erythrozytenreifungsaktivator, ist für MDS mit Ringsideroblasten zugelassen, wenn eine Therapie mit Erythropoetin nicht erfolgreich oder ungeeignet ist. Möglicherweise ist die Wirkung nicht auf MDS mit Ringsideroblasten beschränkt. Die kürzlich präsentierte Phase-III-Studie COMMANDS bei Niedrigrisiko-MDS unabhängig vom Ringsideroblasten-Status zeigte, dass Luspatercept bei ESA(„erythropoiesis-stimulating agent“)-naiven, transfusionsabhängigen MDS-Patient:innen zu 58,5% eine Transfusionsunabhängigkeit (TI) von >12 Wochen und einen Hb-Anstieg von >1,5g/dl erreichte, im Vergleich zuEpoetin alpha mit nur 31,2%; Abb. 1). Die Mortalität war bei beiden Therapien nicht unterschiedlich. Luspatercept könnte aufgrund dieser Studienergebnisse zum neuen Standard in der Erstbehandlung von Anämie bei Niedrigrisiko-MDS werden.
Ein weiteres neues, sehr erfolgversprechendes Medikament für Niedrigrisiko-MDS ist der Telomerase-Inhibitor Imetelstat. In einer kürzlich präsentierten randomisierten, placebokontrollierten, doppelt-blinden Phase-III-Studie (IMerge) bei Niedrigrisiko-MDS-Patient:innen mit Transfusionsbedarf, die gegenüber ESA oder Erythropoeitin (EPO) resistent oder refraktär waren, induzierte Imetelstat gegenüber Placebo eine signifikant höhere Rate an TI >8 Wochen: bei 39,8% der Patient:innen gegenüber 15,0% verglichen mit Placebo.15–18
Therapie des Hochrisiko-MDS
Die allogene Blutstammzelltransplantation stellt die einzig kurative Therapie für Hochrisiko-MDS-Patient:innen dar und ist deswegen die Therapieoption der Wahl. Für Patient:innen, die nicht für eine Transplantation geeignet sind – z.B. wegenhohen Alters (>75 Jahre) oder Begleiterkrankungen – gibt es Azacitidin als Therapiemöglichkeit.
Azacitidin ist ein Cytidin-Analogon, das anstelle von Cytosin in die DNA eingebaut wird. Zusätzlich verhindert es die Methylierung der DNA, indem es das Enzym DNA-Methyltransferase (DNMT) irreversibel bindet. Für Azacitidin ist im Gesamtüberleben ein Vorteil von 6–9 Monaten ausgewiesen (s. randomisierte Studie AZA-001), außerdem eine Verbesserung in Bezug auf Transfusionsfreiheit und periphere Blutwerte gegenüber niedrig dosiertem Cytarabin.
Patient:innen mit Hochrisiko-MDS und CMML mit <13000/μl Leukozyten (dysplastische Variante) können mit Azacitidin behandelt werden, wenn sie nicht für eine allogene Stammzelltransplantation infrage kommen. Da der Effekt der epigenetischen Modulation erst langsam eintritt, sollten mindestens 4–6 Zyklen Azacitidin verabreicht werden, bevor eine Beurteilung des Ansprechens vorgenommen wird. Etwa die Hälfte der Patient:innen erreicht ein Ansprechen mit Verbesserung der peripheren Blutwerte oder einer Remission im KM. Patient:innen, die auf Azacitidin nicht mehr ansprechen, können in Kombination mit Venetoclax behandelt werden („off-label use“). Alternative zu Azacitidin ist Decitabin (i.v. Gabe).19
Literatur:
1 Arber DA et al.: Blood 2022; 140: 1200-82 2 Khoury JD et al.: Leukemia 2022; 36: 1703-19 3 Bhattacharya R et al.: Stroke 2022; 53: 788-97 4 Jaiswal S et al.: N Engl J Med 2014; 371: 2488-98 5 Mas-Peiro S et al.: Clin Res Cardiol 2023; 112: 585-93 6 Mayerhofer E et al.: Stroke 2023; 54: 938-46 7 Bernard E et al.: NEJM Evidence 2022; 1(7) 8 Sauta E et al.: J Clin Oncol 2023; 41: 2827-42 9 List AF et al.: J Clin Oncol 2012; 30: 2134-39 10 Wermke M et al.: Lancet Haematol 2018; 5: e201-10 11 Hellström-Lindberg E et al.: Br J Haematol 1997; 99: 344-51 12 Platzbecker U et al.: Leukemia 2017; 31: 1944-50 13 Fenaux P et al.: Blood 2011; 118: 3765-76 14 López Cadenas F et al.: N Engl J Med 2020; 382: 140-51 16 Garcia-Manero G et al.: J Clin Oncol 2023; 41: 7003 17 Platzbecker U et al.: Lancet 2023; 402: 373-85 18 Platzbecker U et al.: Hemasphere 2023; 7: e0568592 19 Bazinet A et al.: Lancet Haematol 2022; 9: e756
Das könnte Sie auch interessieren:

„Die Lebensqualität ist sehr variabel, auch innerhalb der Subtypen“
Im Rahmen eines JATROS-Interviews mit Ao. Univ.-Prof. Dr. Wolfgang Reinhard Sperr, MedUni Wien, wollten wir einen Einblick in den Umgang mit der seltenen Erkrankung „systemische ...
Anamnese oder präoperatives Gerinnungsscreening?
Blutgerinnungsstörungen können zu intra- wie auch postoperativen Blutungen führen, die abhängig von Lokalisation und Schweregrad ernste Folgen nach sich ziehen können. Ziel einer ...

CAR-T-Zellen: Stellenwert – klinische Evidenz – Herausforderungen
„Chimeric antigen receptor“(CAR)-T-Zellen sind eine der bedeutendsten Innovationen der modernen Hämatologie. Es handelt sich dabei um autologe, genetisch modifizierte T-Zellen, die ex ...