Kutane oder systemische Mastozytose – was macht die Hämatologie?
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Mastzellerkrankungen sind eine heterogene Gruppe von Erkrankungen, die von einer Vielzahl zugrunde liegender genetischer Veränderungen und Komorbiditäten beeinflusst werden und in ihrem klinischen Bild davon geprägt sind. Die Identifikation des Subtyps einer Mastozytose ist das Ziel der hämatologischen Diagnostik, damit eine individuelle Therapie durchgeführt werden kann. Mastzellerkrankungen sind keine allergischen Erkrankungen, obwohl Allergene, aber auch zahlreiche andere Agenzien zu einer Mediatorfreisetzung aus Mastzellen führen können. Dies kann bei der systemischen Mastozytose bis zum anaphylaktischen Schockzustand führen, der lebensbedrohlich sein kann – und daher mit Diagnose und Therapie verhindert werden muss.
Keypoints
-
Diagnostik und Therapie von Mastzellerkrankungen sind ein exquisites Beispiel für Interdisziplinarität – neben der Hausarztpraxis, in der daran gedacht werden muss, braucht es die Dermatologie, die Allergologie, die Pathologie und die Hämatologie.
-
An eine Mastzellerkrankung zu denken, führt neben einer ausführlichen Anamnese und klinischen Untersuchung zur ersten diagnostischen Analyse: der Bestimmung der Serum-Tryptase. Dies kann bereits in der Hausarztpraxis erfolgen.
-
Die kutane muss von der systemischen Mastozytose unterschieden werden, da Prognose und therapeutische Optionen unterschiedlich sind. Diese Unterscheidung ist ohne eine Knochenmarkuntersuchung nicht möglich.
-
Sowohl diagnostisch (Nachweis aktivierender KIT-Mutationen und einer aberranten Expression von CD2, CD25 und/oder CD30 auf neoplastischen Mastzellen) als auch therapeutisch (Einsatz von KIT-Inhibitoren) sind in den letzten Jahren relevante Fortschritte bei den Mastzellerkrankungen erzielt worden.
-
Die Differenzialdiagnose ist breit und umfasst insbesondere die hereditäre Alpha-Tryptasämie als mit Abstand häufigsten Grund für eine erhöhte basale Serum-Tryptase, das Mastzellaktivierungs-Syndrom und die Histaminintoleranz. Da auch hier Prognose und therapeutische Interventionsmöglichkeiten verschieden sind, ist eine diagnostische Zuordnung wichtig.
Am 27. März 2025 hat ein vom Luzerner Kantonsspital organisierter Workshop zur Mastozytose – «Diagnostik und Therapie von Mastzellerkrankungen» – stattgefunden.
Die dabei notwendige Interdisziplinarität spiegelte sich in drei Referaten wider:
Dr. med. Anna Kirsch vom Zentrum für Dermatologie und Allergologie am Luzerner Kantonsspital beleuchtete in ihrem Referat die dermatologische Problematik, PD Dr. med. Oliver Fuchs, ebenfalls vom Zentrum für Dermatologie und Allergologie, thematisierte die allergologischen Abklärungen inklusive der Bedeutung der Serum-Tryptase im Basalzustand und im Anfall einer Mastzelldegranulation, und KD Dr.med. Axel Rüfer referierte zum Thema der Unterscheidung von kutaner und systemischer Mastozytose und zu den Aufgaben der Hämatologie dabei, inklusive klinisch relevanter Differenzialdiagnosen.
Das Luzerner Kantonsspital ist ein Exzellenzzentrum des European Competence Network on Mastocytosis (ECNM). Im folgenden Artikel geht es um die genuin hämatologischen Aspekte der Mastozytose und ihre Differenzialdiagnosen im klinischen Alltag.
Kutane oder systemische Mastozytose?
Während die kutane Mastozytose (CM) überwiegend Kinder betrifft, ist die systemische Mastozytose (SM) in der Regel eine Erkrankung von Erwachsenen. Auch bei Letzteren können typische Hautläsionen mit Mastzellinfiltraten auftreten und eine Knochenmarkuntersuchung zum Staging ist unverzichtbar.
Die Unterscheidung zwischen CM und SM ist prognostisch wichtig, da das progressionsfreie Überleben bei der CM länger ist als bei der SM. Die Abbildung 1 zeigt für Mastzellinfiltrate typische Hautläsionen, durch mechanisches Reiben einer solchen Läsion lässt sich bei Patient:innen, die kein Antihistaminikum einnehmen, in der Regel eine Quaddel auslösen (positives Darier-Zeichen) (Abb. 1).
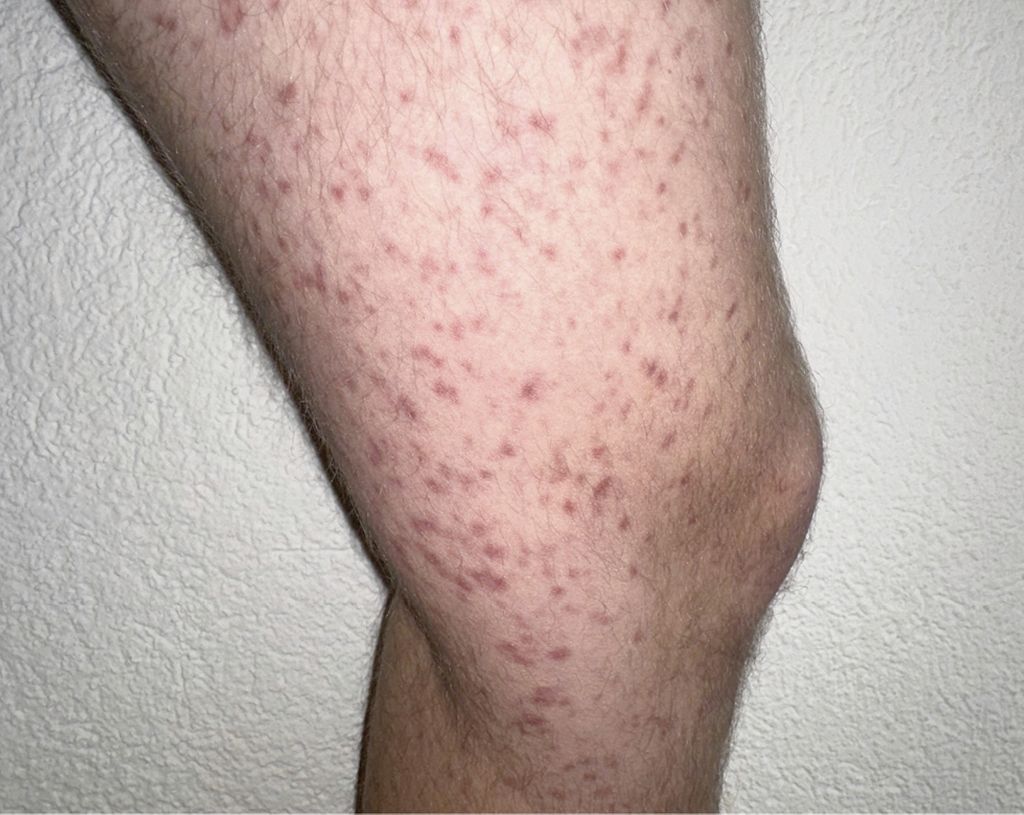
Abb.1: Kutane oder systemische Mastozytose – bei für Mastzellinfiltrate typischen Hautläsionen? Mit freundlicher Genehmigung.
Die SM ist durch eine pathologische Vermehrung neoplastischer Mastzellen im Knochenmark und in anderen Organen charakterisiert, vor allem in Milz, Leber und Lymphknoten. Mastzellen exprimieren die Rezeptor-Tyrosinkinase KIT (CD117) und in >90% der Patient:innen mit SM ist eine aktivierende KIT-Mutation nachweisbar, die zu einer Aktivierung des KIT-Rezeptors mit klonaler Expansion und Akkumulation von Mastzellen in oben erwähnten Organen führt.1
Wann an Mastozytose denken? – Symptome und Zeichen
Die klinischen Zeichen einer Mastzellerkrankung können mannigfaltig sein, sind unspezifisch und hängen davon ab, ob eine nichtfortgeschrittene oder eine fortgeschrittene SM vorliegt.
Bei der nichtfortgeschrittenen SM treten neben konstitutionellen Symptomen (Fatigue, Gewichtsverlust, Fieber) und Hautmanifestationen (Urtikaria, Angioödeme, Dermographismus, Juckreiz, Flush) überwiegend mediatorassoziierte systemische Symptome auf (gastrointestinal mit Übelkeit, Erbrechen und Nahrungsmittelunverträglichkeiten, respiratorisch mit Atemnot und Stridor, Herz/Kreislauf mit Hypotonie, Tachykardie, Synkope, Schwindel), und muskuloskelettale Symptome (Knochenschmerzen, Arthralgien, Myalgien) sind häufig. Die klassische klinische Präsentation ist eine höhergradige anaphylaktische Reaktion (Klassifikation nach Müller, Grad 3: Atemnot, oder Grad 4: anaphylaktischer Schock) nach einem Hymenopterenstich – bei einer Allergie auf Hymenopterengifte von Biene, Wespe oder Hornisse.
Der Phänotyp bei der fortgeschrittenen SM ist gekennzeichnet durch die Vergrösserung und Funktionseinschränkung von Organen durch die Mastzellinfiltration, wobei Knochenmark, Milz, Leber, Gastrointestinaltrakt, Lymphknoten und/oder Knochen betroffen sein können.
Wie so oft ist es daher wichtig, an eine Mastzellerkrankung zu denken, um den weiteren Weg für Diagnostik und Therapie zu bereiten, wobei oft zuerst der Hausarzt/die Hausärztin kontaktiert werden.2 Auch bei anaphylaktischen Reaktionen ohne eine Hautbeteiligung muss an eine (Knochenmark-)Mastozytose («bone marrow mastocytosis» [BMM]) gedacht werden.
Diagnostische Kriterien für systemische Mastozytose
In der Hausarztpraxis soll bei einer derartigen klinischen Präsentation die Serum-Tryptase bestimmt werden, die eine sehr hohe Spezifität aufweist. Dabei ist wichtig, den Zeitpunkt nach einem Ereignis zu dokumentieren. Es gibt einen Peak der Serum-Tryptase 30 bis 120 Minuten nach Auftreten von Symptomen. Eine basale Serum-Tryptase (BST) sollte mehr als 24 Stunden nach Abklingen der Symptome bestimmt werden bzw. im symptomfreien Intervall.
Sollte eine erhöhte BST vorliegen, ist der häufigste Grund dafür eine hereditäre Alpha-Tryptasämie, gefolgt von einer fortgeschrittenen chronischen Nierenerkrankung/terminalen Niereninsuffizienz und einer Mastozytose.3 Da unstimulierte Mastzellen eine minimale Menge an (Pro-)Tryptase freisetzen, ist die BST Ausdruck der Gesamtzahl von Mastzellen.
Für die Diagnose SM gibt es entsprechend der WHO-Klassifikation 2022 ein Hauptkriterium und vier Nebenkriterien.4 Die diagnostischen Kriterien sind in Tabelle 1 dargestellt. Da die CD30-Expression von Mastzellen assoziiert ist mit systemischer Mastozytose, wurde diese dem entsprechenden Nebenkriterium hinzugefügt. Zudem wurde die Existenz einer anderen aktivierenden KIT-Mutation (nicht ausschliesslich dieKIT-p.D816V-Mutation) als Nebenkriterium anerkannt. Die Diagnose einer SM kann gestellt werden, wenn das Hauptkriterium und mindestens ein Nebenkriterium oder mindestens drei Nebenkriterien erfüllt werden.

Tab.1: Diagnostische Kriterien für eine systemische Mastozytose
In einer retrospektiven Analyse des ECNM-Registers konnte gezeigt werden, dass die aberrante Expression von Mastzellen auch prognostische Wertigkeit hat und dass eine fehlende CD2-Expression mit einem signifikant reduzierten Gesamtüberleben und einer signifikant gesteigerten extramedullären Beteiligung assoziiert ist.5
Es sei darauf hingewiesen, dass ein BST-Normalwert oder eine nur leicht erhöhte BST <20ng/ml eine Mastozytose nicht ausschliesst und dass die Serum-Tryptase gelegentlich bei schweren Anaphylaxien nur minimal erhöht sein kann.
Verdacht auf systemische Mastozytose: hämatologische Diagnostik
Nachdem aufgrund der Anamnese, der klinischen Symptome und der körperlichen Untersuchung sowie der Bestimmung der BST der Verdacht auf eine Mastozytose geäussert worden ist, ist eine hämatologische Diagnostik indiziert. Diese beinhaltet zunächst eine Differenzierung des Blutbildes (Morphologie der Monozyten und Eosinophilen, Dysplasien) und die Bestimmung von Laborparametern (Serum-Tryptase, Transaminasen, AP, LDH, Bilirubin, Kreatinin, Albumin, Ferritin, Vitamin B12, Erythrozyten-Folsäure, Kalzium, Vitamin D, CRP, Serum-Proteinelektrophorese mit Immunfixation, plasmatische Gerinnung).
Mit dem Nachweis einer KIT-p.D816V-Mutation durch eine ultrasensitive «Droplet digital»-PCR aus dem peripheren Blut lässt sich der Verdacht auf eine Mastozytose weiter erhärten und die Notwendigkeit einer Knochenmarkuntersuchung durch eine erhöhte Vortest-Wahrscheinlichkeit noch besser belegen.
Diese KIT-p.D816V-Mutation ist bei ungefähr 95% der erwachsenen Patient:innen mit einer nichtfortgeschrittenen SM vorhanden. Es ist dabei zu beachten, dass sowohl BMM als auch indolente SM (ISM) bei einer geringen Mastzelllast im peripheren Blut negativ, im Knochenmark positiv sein können. Zudem können andere KIT-aktivierende und somit krankheitstreibende Mutationen (z.B. D816Y, D816H, D816F, D815K, F522C, V560G, D820G) vorliegen, insbesondere bei fortgeschrittener SM. Hier sind neben der KIT-p.D816V-Mutation zusätzliche, rekurrente Mutationen nachweisbar. Am häufigsten betroffen sind SRSF2, ASXL1 und RUNX1 (S/A/R-Gen-Panel), die für die Risikostratifizierung im molekularen MARS-Score bedeutsam sind.6
Mit einer Knochenmarkuntersuchung wird ein Aspirat für Zytologie, Immunphänotypisierung (FACS) und genetische Untersuchungen gewonnen sowie eine Biopsie für Histologie, Immunhistochemie und genetische Untersuchungen. Eine Prämedikation zur Prävention von Mastzelldegranulationen bei dieser Untersuchung ist nicht notwendig.
Bei der Zytologie werden die Zellularität und allfällige Dysplasien beurteilt. Zudem ist eine Quantifizierung der Mastzellen wichtig, da ≥20% ausserhalb der Bröckel als ein Kriterium für eine Mastzell-Leukämie (MCL) gelten. Zudem wird am Knochenmarkaspirat ein FACS durchgeführt. Genetische Untersuchungen beinhalten die molekulargenetische Bestimmung der KIT-p.D816V-Mutation mit Mutationslast («variant allele frequency» [VAF]) und eine konventionelle zytogenetische Chromosomenanalyse. Allerdings gibt es keine spezifischen Aberrationen bei der SM, zumal diese bei der BMM und der ISM selten sind.
In der Biopsie wird neben der Quantifizierung der Mastzellen vor allem beurteilt, ob multifokale, dichte Infiltrate von Mastzellen als diagnostisches Hauptkriterium vorliegen. Neben einer Faserfärbung ist die Immunhistochemie wichtig zur Beurteilung der Mastzellen (Tryptase, CD117, CD2, CD25, CD30) und einer allfälligen assoziierten hämatologischen Neoplasie (CD14, CD34, CD61).
Systemische Mastozytose –aber welcher Subtyp?
Kann durch die hämatologische Diagnostik die Diagnose SM gestellt werden, ist die nächste Frage, welcher Subtyp vorliegt. Dies hat nicht nur prognostische, sondern auch therapeutische Konsequenzen. Um den Subtyp festzulegen, ist es wichtig, das Vorhandensein von «B findings» («burden of disease») oder «C findings» («cytoreduction required») zu prüfen. «C findings» sind letztlich durch Mastzellinfiltration bedingte Funktionseinschränkungen von betroffenen Organen. «B findings» und «C findings» sind in Tabelle 2 zusammengefasst.

Tab.2: «B findings» und «C findings»
Um die diagnostischen Abklärungen abzuschliessen, muss eine Sonografie des Abdomens mit der Frage nach Hepato-/Splenomegalie, Lymphadenopathie, Aszites und portaler Hypertension erfolgen.
Zudem muss bei diesen Patient:innen eine Osteodensitometrie vorgenommen werden – die nicht kassenpflichtig ist –, mit der Frage, ob eine Osteopenie oder Osteoporose vorliegt. Dies ist für die Diagnose und auch für die Therapie wichtig. Die Serum-Tryptase induziert, zusammen mit anderen Zytokinen der Mastzellen, die Bildung von RANKL («receptor activator of NF-κB ligand») in Osteozyten und Osteoblasten und stimuliert so die Osteoklasten, wobei insbesondere die Wirbelsäule betroffen ist. Bei Verdacht auf Osteolysen muss eine Magnetresonanztomografie durchgeführt werden.
Bei gastrointestinalen Symptomen sollten eine Ösophagogastroduodenoskopie und Koloskopie erfolgen. Dabei sollte immer eine Biopsie mit CD117-Immunhistochemie vorgenommen werden, da Mastzellen immer CD117 exprimieren, die Tryptase hingegen fehlen kann. Bei Verdacht auf gastrointestinale Mastzellinfiltration muss die Pathologie über diesen Verdacht informiert werden, um die entsprechende Immunhistochemie an den Biopsaten durchzuführen und um die Fehldiagnose einer eosinophilen Kolitis zu vermeiden.
In Kenntnis aller dieser Parameter kann nun der Subtyp der SM festgelegt werden. Zu den nichtfortgeschrittenen SM gehören die BMM, die ISM und die «smouldering» SM (SSM). Bei der BMM gibt es keine Hautläsionen und keine «B findings», die BST ist <125ng/ml und es gibt keine dichten extramedullären SM-Infiltrate. Eine ISM liegt vor bei typischen Hautläsionen und maximal einem «B finding». Es existiert die ISM auch ohne Hautläsionen, allerdings ist dann die BST ≥125ng/ml und/oder es gibt dichte extramedulläre SM-Infiltrate. Bei der SSM gibt es mindestens zwei «B findings», aber keine «C findings».
Die fortgeschrittenen SM (AdvSM) sind die aggressive SM (ASM), die SM mit assoziierter hämatologischer Neoplasie (AHN) und die MCL. Bei der ASM gibt es mindestens ein «C finding» und oft fehlen Hautläsionen. Bei der SM-AHN muss eine WHO-definierte hämatologische Neoplasie zusätzlich zur SM vorliegen, beide Erkrankungen werden klassifiziert nach der Definition der WHO. Bei der MCL gibt es ≥20% Mastzellen im Knochenmark-Ausstrich, bei der leukämischen Form sind ≥10% Mastzellen im peripheren Blut, bei der aleukämischen Variante sind es <10%. Liegt eine akute MCL vor, sind ein oder mehrere «C findings» vorhanden, während es bei der chronischen MCL, die eine deutlich bessere Prognose hat, keine «C findings» gibt.
Therapie dersystemischen Mastozytose
Die therapeutischen Optionen bei der SM sind in der Abbildung 2 zusammengefasst. Zulassungsstatus, Verfügbarkeit und Kostenübernahme werden bei dieser Darstellung nicht berücksichtigt und müssen vor der Therapie geklärt werden.
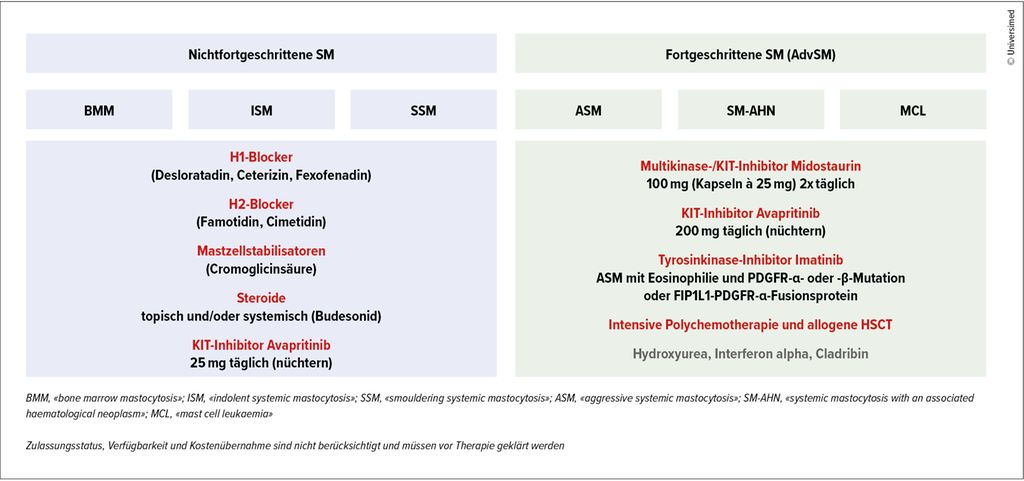
Abb. 2: Therapeutische Optionen bei verschiedenen Subtypen der systemischen Mastozytose
Da der klinische Verlauf der nichtfortgeschrittenen SM individuell sehr verschieden ist, ist es auch die Therapie. Diese beginnt immer mit der Vermeidung potenziell auslösender Agenzien, wobei zur Identifikation das Führen eines Tagebuchs empfohlen wird. In der Regel wird eine prophylaktische Anti-Mediator-Therapie durchgeführt, mit der Einnahme von H1-Blockern (z.B. Desloratadin, Ceterizin, Fexofenadin u.a.) und dem (abendlichen) Einsatz von H2-Blockern (z.B. Famotidin 2–3x40mg/d oder Cimetidin 2–3x 400mg/d) bei gastrointestinalen Beschwerden. Sollten diese mit einem H2-Blocker nicht zufriedenstellend kontrolliert werden, kann zusätzlich – zeitversetzt – ein Protonenpumpen-Inhibitor eingesetzt werden. Eine Therapieakzentuierung kann mit dem Einsatz eines Mastzellstabilisators (z.B. Cromoglicinsäure) oder von Steroiden (z.B. Budesonid) erreicht werden.
Bei einer Hymenopterengiftallergie sollte eine spezifische Immuntherapie als lebenslange Dauertherapie erfolgen. Liegen mittelschwere bis schwere Symptome vor, bei denen mit dieser symptomatischen Therapie keine ausreichende Kontrolle erfolgt, kann die Therapie der ISM mit dem spezifischen KIT-Inhibitor Avapritinib erfolgen, in einer täglichen Dosis von 25mg.7
Die Therapie der fortgeschrittenen SM differiert im Vergleich zu jener der nichtfortgeschrittenen SM. Für die Erstlinientherapie der fortgeschrittenen SM wird der Multikinase-/KIT-Inhibitor Midostaurin in einer Dosis von 2x100mg (Kapseln à 25mg) täglich eingesetzt, wobei Übelkeit ein häufiges Problem darstellt, sodass eine regelmässige Prophylaxe z.B. mit Ondansetron 4–8mg/d erfolgen muss. Der spezifische KIT-Inhibitor Avapritinib ist als Monotherapie in einer Dosis von 200mg täglich nach mindestens einer systemischen Vortherapie zugelassen, wobei die Thrombozyten >50G/L betragen müssen.8,9
Eine intensive Polychemotherapie in Kombination mit Midostaurin, gefolgt von einer allogenen hämatopoetischen Stammzelltransplantation (HSCT), ist bei rasch progredienter ASM, SM-AML und MCL notwendig. Eine posttransplantäre Therapie mit einem KIT-Inhibitor soll dann in Erwägung gezogen werden.
Hydroxyurea kann im palliativen Setting zum Einsatz kommen, insbesondere bei symptomatischer Leukostase oder Splenomegalie. Interferon alpha und Cladribin sind seit der Verfügbarkeit von Midostaurin und Avapritinib in den Hintergrund getreten.
Sowohl eine Osteopenie als auch eine Osteoporose müssen adäquat behandelt und weitere sekundäre Ursachen, wie Vitamin-D3-Mangel, Hyperthyreose, Hyperparathyreoidismus oder Hypogonadismus, ausgeschlossen werden. Zudem muss eine tägliche Einnahme von 1000–1200mg Kalzium und 800–1200 Einheiten Vitamin D sichergestellt sein. Eine Bisphosphonat-Therapie – nach zahnärztlicher Kontrolle – sollte eingeleitet werden, sobald der T-Score unter –2 abfällt, um eine sich schnell verschlechternde Osteoporose frühzeitig aufzuhalten. Ist eine Stabilisierung der Osteoporose nicht möglich, muss der Einsatz des RANKL-Inhibitors Denosumab diskutiert werden. Allerdings muss eine solche Therapie gefolgt werden von einem Bisphosphonat, um einen Knochenumbau-Rebound zu verhindern. Teriparatid ist im Gegensatz zu Bisphosphonaten knochenanabol wirksam und ist bis jetzt das am besten wirksame Präparat, insbesondere bei schwerer, manifester Osteoporose oder bei trotz Bisphosphonattherapie aufgetretenen Frakturen.
Zur Therapie einer höhergradigen Anaphylaxie (Grad 3 oder Grad 4, Klassifikation nach Müller) als Notfall müssen alle Patient:innen mit SM mit einem Notfallset ausgestattet sein, in dem jeweils zwei Tabletten mit einem H1-Blocker (z.B. Levoceterizin, 5mg) und mit Prednisolon (z.B. Spiricort, 50mg) enthalten sind. Zudem ist es wichtig, solche Patient:innen mit zwei Auto-Adrenalin-Injektoren mit jeweils 0,3mg Adrenalin auszustatten, die im Notfall selbstständig intramuskulär verabreicht werden sollen.
Alle Patient:innen mit einer Mastozytose sollten nach Möglichkeit in Studien behandelt werden. Auch sollte ein Einschluss in ein internationales Mastozytose-Register erfolgen (z.B. ECNM-Register), das die Grundlage für Datenanalysen ist.
Klinisch relevante Differenzialdiagnosen
Hereditäre Alpha-Tryptasämie
Bei Symptomen, die für eine Mediatorenfreisetzung aus den Mastzellen sprechen, muss bei erhöhten Werten der Serum-Tryptase an die hereditäre Alpha-Tryptasämie (HAT) gedacht werden. Dies ist auch bei gastrointestinalen Symptomen mit nächtlichem Erwachen oder Darmentzündungen, die nicht auf die Therapie ansprechen, der Fall. Die HAT ist der häufigste Grund für eine erhöhte BST.10 Sie findet sich mit einer Prävalenz von 4–6% in der westlichen Bevölkerung und ist ein autosomal-dominant vererbtes Merkmal, das durch vermehrte Keimbahnkopien des Alpha-Tryptase-codierenden TPSAB1-Gens verursacht wird.
Bei BST-Werten ≥8ng/ml soll bei diesen symptomatischen Patient:innen die HAT gesucht werden. Die HAT ist bei Patient:innen mit SM gehäuft. Die Therapie der HAT ist ähnlich wie bei der SM, und die Basistherapie sind Antihistaminika. Kurzfristig können zur Symptomkontrolle auch H2-Blocker, Leukotrien-Antagonisten und Cromoglicinsäure eingesetzt werden; es gibt einzelne Fallberichte einer erfolgreichen Urtikariabehandlung mit Omalizumab. In Notfallsituationen sind in der Regel Antihistaminika und Steroide ausreichend.11
Mastzellaktivierungs-Syndrom
Symptome der Mastzellaktivierung (MCA) sind vielfältig, wie z.B. Urtikaria, Angioödem, Pruritus, Flush, verstopfte Nase, pfeifende Atmung («Wheezing»), Kehlkopfschwellung, Kopfschmerzen, Hypotonie oder Diarrhö. Bei Patient:innen mit schweren Symptomen einer MCA, die systemisch sind, mehr als ein Organsystem betreffen und wiederholt auftreten, in der Regel in Form einer schweren Anaphylaxie, muss an ein Mastzellaktivierungs-Syndrom (MCAS) gedacht werden. Dies gilt als ein Diagnosekriterium.
Eine normale BST schliesst ein MCAS nicht aus. Es muss eine Erhöhung der Serum-Tryptase um mindestens 20%+2ng/ml über der BST innerhalb von vier Stunden nach Auftreten von klinischen Symptomen dokumentiert werden, um von einer MCA ausgehen zu können (Beispiel: BST 10ng/ml → Serum-Tryptase «im Anfall» muss >14ng/ml sein, um als MCA-Kriterium zu gelten). Dieser Anstieg der Serum-Tryptase gilt als ein weiteres Diagnosekriterium. Ausserdem muss es ein Therapieansprechen geben, wobei die therapeutische Intervention personalisiert sein muss und abhängig von der zugrunde liegenden Ätiologie ist (Mastzellreduktion, Mastzellstabilisierung, Anti-Mediator-Therapie, Anti-Allergie-Therapie, Immuntherapie, Omalizumab, Steroide, Antiinfektiva, symptomatische Therapie). Auch das Therapieansprechen ist ein Diagnosekriterium. Für die Diagnose eines MCAS müssen alle drei diagnostischen Kriterien erfüllt sein.12
Es gibt drei Varianten des MCAS, die für die therapeutische Intervention ausschlaggebend sind:
-
das primäre (klonale=monoklonale) MCAS, bei dem klonale Mastzellen detektiert werden können, entweder mit der Diagnose Mastozytose (CM oder SM) oder mit nur zwei Nebenkriterien für die Diagnose SM,
-
das sekundäre (reaktive) MCAS, das durch eine IgE-vermittelte Allergie, eine andere Hypersensitivitätsreaktion oder eine andere immunologische Erkrankung verursacht werden kann, und
-
das idiopathische MCAS, bei dem es keine assoziierte reaktive Erkrankung, keine IgE-vermittelte Allergie, keine HAT und keine klonalen Mastzellen gibt.
HAT und MCAS können auch gemeinsam auftreten, dies gilt auch für das primäre und sekundäre MCAS. Es sei darauf hingewiesen, dass ein MCAS Ausdruck einer anderen Mastzellerkrankung oder einer IgE-vermittelten Erkrankung sein kann.
Histaminintoleranz
Die sogenannte Histaminintoleranz ist laut der Internationalen Klassifikation der Krankheiten keine offizielle Diagnose. Einige Patient:innen können auf die zusätzliche Histaminaufnahme mit einer Vielzahl von unspezifischen Symptomen im Sinne einer Nahrungsmittelunverträglichkeit reagieren, z.B. mit Bauchschmerzen, Durchfall, Übelkeit, Erbrechen, Hypotonie, Tachykardie, Schwindel, Niesen, verstopfter Nase, Quaddelbildung, Juckreiz oder Flush. Nahrungsmittel mit besonders viel Histamin sind bestimmte (lang gereifte) Käsesorten, Fleischsorten (Salami, Rohschinken), Fischsorten (Konserven, Thunfisch, Makrele), Alkohol (Rotwein, Champagner), aber auch Tomaten, Trauben, Erdbeeren oder Nüsse. Möglicherweise liegt eine Dysbalance zwischen Aufnahme und Abbau von Histamin vor. Das Führen eines Symptom- und Ernährungstagebuchs ist hilfreich bei Patient:innen mit Verdacht auf Histaminintoleranz.
Für die Diagnosestellung gibt es keinen einzelnen Labortest. Die Bestimmung der Aktivität der Diaminoxidase (DAO) im Serum hat keine diagnostische Aussagekraft, da sie keinen Rückschluss auf die Enzymaktivität im Dünndarm zulässt. Zudem erfolgt der Abbau von Histamin im Darm möglicherweise auch durch die Histamin-N-Methyl-Transferase. Die Bestimmung der Histaminkonzentration im Plasma oder die Bestimmung von Histamin im Stuhl ist in ihrer diagnostischen Wertigkeit umstritten, insbesondere da Darmbakterien Histamin als relevantes Stoffwechselprodukt haben.
Differenzialdiagnostisch müssen sowohl eine Mastzellerkrankung als auch eine IgE-vermittelte Allergie ausgeschlossen werden.
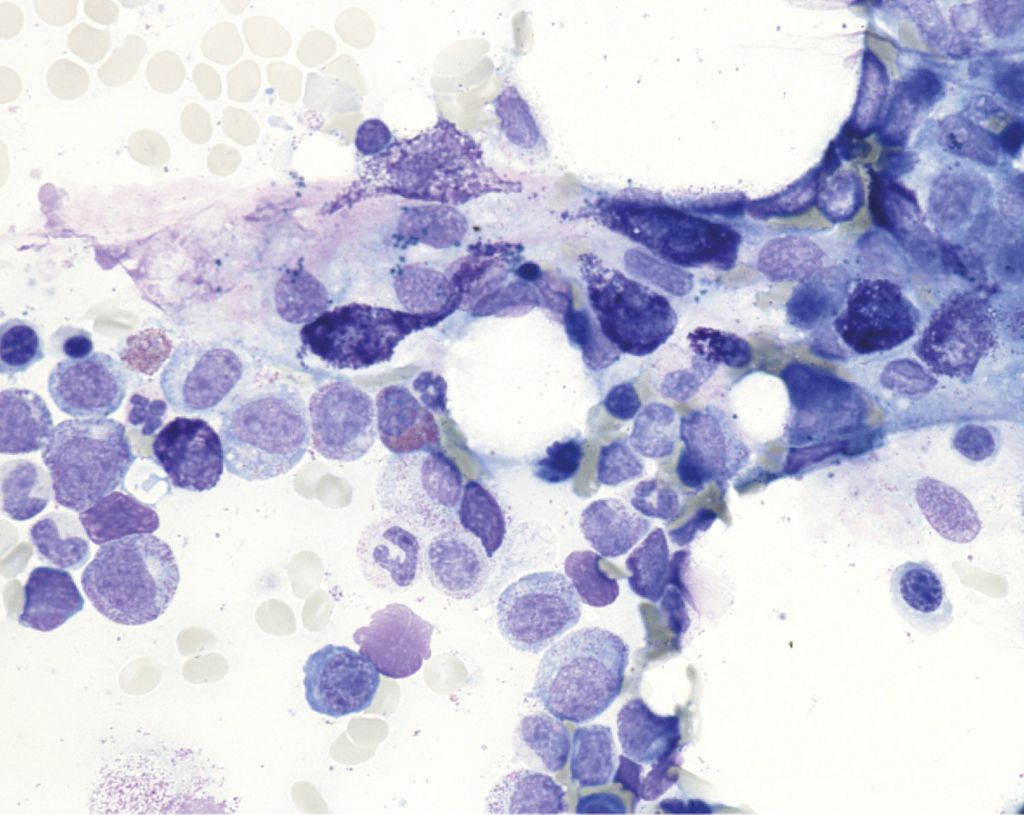
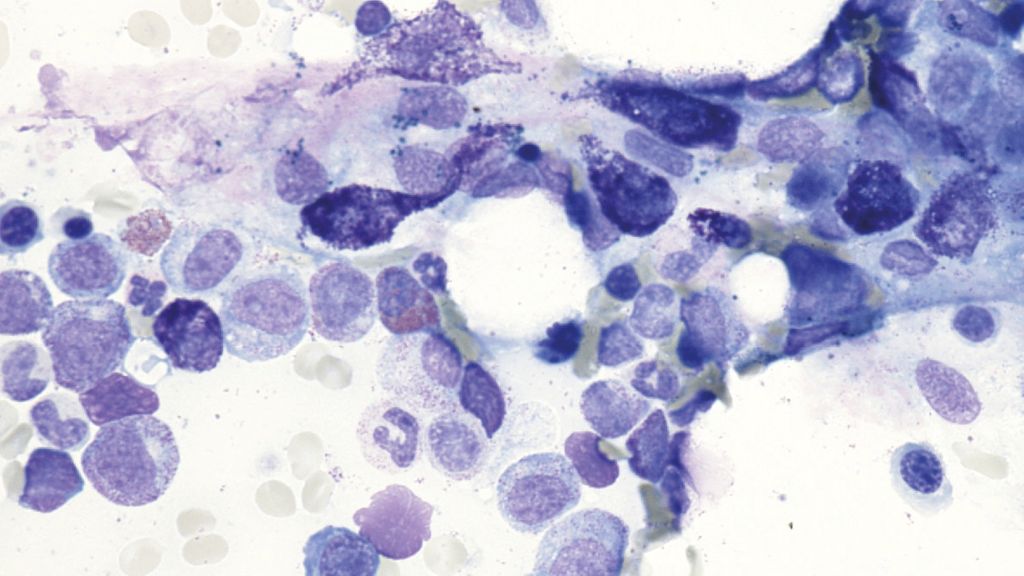
Abb. 3: Knochenmark-Aspirat
Zudem muss in der ersten Phase der Ernährungsumstellung – der Karenzphase – eine Ernährungsberatung stattfinden und für 2–4 Wochen eine histaminarme Kost aufgenommen werden. Führt diese Karenzphase zu einer Symptombesserung, ist eine Histaminintoleranz sehr wahrscheinlich, und in einer zweiten Phase – der Testphase – ist es das Ziel, die Nahrungsmittelauswahl durch gezielte Wiedereinführung verdächtiger Nahrungsmittel unter Berücksichtigung individueller Einflussfaktoren zu erweitern. Die dreistufige Ernährungsumstellung kulminiert in der dritten Phase – der Dauerernährung – mit dem Ziel einer langfristigen, individuellen symptomorientierten Ernährungsempfehlung zur bedarfsdeckenden Nährstoffzufuhr und zur Vorbeugung der Entwicklung von Mangelerscheinungen bei hoher Lebensqualität.
Neben dem Vermeiden von Symptomauslösern kommen auch Antihistaminika oder das Nahrungsergänzungsmittel DAO zum Einsatz, das zu einem beschleunigten Histaminabbau führt und vor dem Essen eingenommen werden sollte. Wissenschaftlich belegt ist die Wirksamkeit einer Supplementation von DAO bisher allerdings nicht.
Wenn die Ernährungsberatung und Aufnahme histaminarmer Kost nicht zu einer Verbesserung der Symptome führen, empfiehlt sich eine gastroenterologische Diagnostik (Differenzialdiagnosen Reizdarmsyndrom/Colon irritabile, chronisch-entzündliche Darmerkrankungen, Zöliakie, Ulcus ventriculi/duodeni).13,14
Eine gestörte Barriere der Darmschleimhaut, z.B. durch Entzündungen, kann u.a. zu einer erhöhten Ausschüttung von Histamin führen, und es gibt Einzelfallberichte des erfolgreichen Einsatzes von Vitamin A und D, Zink, kurzkettigen Fettsäuren, Methionin, Glutamin und Probiotika in einer solchen Situation – allerdings fehlen randomisierte, kontrollierte Studien.
Bei der klinischen Symptomatik gibt es viele Überlappungen von CM und SM, HAT, MCAS und Histaminintoleranz. Emotionaler Stress kann zur Auslösung von Mastzelldegranulationen führen und Symptome bei Mastozytose, HAT, MCAS und Histaminintoleranz entweder auslösen oder zumindest verstärken. Es ist daher unabdingbar, neben den oben beschriebenen therapeutischen Optionen auch eine psychologische Beratung anzubieten und durchführen zu lassen.
Literatur:
1 Valent P et al.: Updated diagnostic criteria and classification of mast cell disorders: a consensus proposal. Hemasphere 2021; 5(11): e646 2 Theoharides TC et al.: Mast cells, mastocytosis, and related disorders. N Engl J Med 2015; 373(2): 163-72 3 Waters AM et al.: Elevated basal serum-tryptase: disease distribution and variability in a regional health system. J Allergy Clin Immunol Pract 2022; 10(9): 2424-35 4 The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36(7): 1703-19 5 Rüfer A et al.: Prognostic impact of expression of CD2, CD25, and/or CD30 in/on mast cells in systemic mastocytosis: a registry study of the European Competence Network on Mastocytosis. Leukemia 2025; 39(3): 675-83 6 Jawhar M et al.: MARS: Mutation-adjusted risk score for advanced systemic mastocytosis. JClin Oncol 2019; 37(31): 2846-56 7 Gotlib J et al.: Avapritinib versus placebo in indolent systemic mastocytosis. N Engl J med Evid 2023; 2(6): EVIDoa2200339 8 DeAngelo DJ et al.: Safety and efficacy of avapritinib in advanced systemic mastocytosis: the phase 1 EXPLORER trial. Nat Med 2021; 27: 2183-91 9 Gotlib J et al.: Efficacy and safety of avapritinib in advanced systemic mastocytosis: interim analysis of the phase 2 PATHFINDER trial. Nat Med 2021; 27(12): 2192-9 10 Lyons JJ et al.: Incorporating tryptase genotyping into the workup and diagnosis of mast cell diseases and reactions. J Allergy Clin Immunol Pract 2022; 10(8): 1964-73 11 Rüfer A et al.: Hereditary alpha-tryptasemia – a potential cause of severe anaphylactic reactions and a modifier of mast cell diseases. Swiss Med Wkly 2025; 155: 3679-84 12 Valent P et al.: Mast cell activation syndromes: Collegium Internationale Allergologicum update 2022. Int Arch Allergy Immunol 2022; 183(7): 693-705 13 Hrubisko M et al.: Histamine intolerance – the more we know the less we know. A review. Nutrients 2021; 13(7): 2228 14 Reese I et al.: Guideline on management of suspected adverse reactions to ingested histamine: Guideline of the German Society for Allergology and Clinical Immunology (DGAKI), the Society for Pediatric Allergology and Environmental Medicine (GPA), the Medical Association for German Allergologists (AeDA) as well as the Swiss Society for Allergology and Immunology (SGAI) and the Austrian Society for Allergology and Immunology (ÖGAI). Allergol Select 2021; 5: 305-14
Das könnte Sie auch interessieren:
Therapie im Wandel: zeitlich begrenzt und Sequenzierung
Die therapeutische Landschaft der chronischen lymphatischen Leukämie (CLL) hat sich in den vergangenen Jahren fundamental gewandelt. Die klassische Chemoimmun-therapie ist weitgehend ...
Chronische myeloische Leukämie (CML)
Die Prognose der CML-Patient:innen unterscheidet sich durch die Entwicklung der Tyrosinkinaseinhibitoren (TKI) kaum mehr von der Normalbevölkerung. Dennoch gibt es neue Erkenntnisse, die ...
Therapieneuerungen: FL, CLL und LBCL
Am Jahreskongress der American Society of Hematology (ASH) wurden zahlreiche relevante Neuerungen in der Lymphomtherapie vorgestellt. Wir präsentieren drei ausgewählte Studien zur ...