Langerhanszell-Histiozytose: Trials und Register im Erwachsenenalter
Histiozytäre Erkrankungen sind eine Rarität im Erwachsenenalter. Mit einer Inzidenz von circa 1/1000000 stellt die Langerhanszell-Histiozytose (LCH) die häufigste Subentität dar. Andere Formen wie Erdheim-Chester- oder Rosai-Dorfmann-Disease sind noch seltener.1,2 Wie bei vielen «orphandiseases» führt dies dazu, dasses an einerStandardisierung der Diagnostik und Therapie dieser zum Teil schwer erkrankten Patienten mangelt. Im Rahmen der Session «Langerhanszell-Histiozytose» während der DGHO-Jahrestagung 2020 wurden Herausforderungen und Lösungsansätze im Management der LCH diskutiert.
Keypoints
-
Die Langerhanszell-Histiozytose ist eine seltene Erkrankung im Erwachsenenalter. Das klinische Erscheinungsbild ist sehr variabel.
-
Der Differenzierungsgrad der Ursprungszelle entscheidet das klinische Bild der Langerhanszell-Histiozytose.
-
Alterationen des MAPK-Signalwegs spielen eine wichtige Rolle in der Entstehung der LCH und können therapeutisch mittels BRAF- und MEK-Inhibitoren adressiert werden.
-
Die Haut ist am zweithäufigsten durch die LCH betroffen. Eine dermatologische Vorstellung sollte daher Teil jedes diagnostischen Work-ups sein.
-
Während die Therapie bei Kinder gut etabliert ist, fehlen für erwachsene Patienten randomisierte Studien. Einen ersten Schritt in diese Richtung stellt das neu etablierte Züricher Histiozytose-Netzwerk am Universitätsspital Zürich dar.
Die LCH – eine entzündliche myeloische Neoplasie
Dr. med. Matthias Wilk, PhD (Icahn School of Medicine, Mount Sinai, New York), begann die Session mit einem Überblick über die aktuelle Forschung zur Pathogenese histiozytärer Erkrankungen. Charakteristisch ist eine Akkumulation von phagozytischen Zellen. Diese Infiltrationen können in den unterschiedlichsten Geweben auftreten. Aufgrund des breiten klinischen Bildes – von sehr milden, lokalisierten Erkrankungen bis hin zu lebensbedrohlichen Verläufen mit Multiorganbefall – ist die Pathophysiologie dieser Erkrankung Gegenstand anhaltender Diskussion. Dies, zumal histiozytäre Erkrankungen sowohl Merkmale einer entzündlichenals auch einer neoplastischen Erkrankung zeigen. Forschungsergebnisse der letzten 10 Jahre konnten aktivierende Mutationen im MAPK-Signalweg als relevantes gemeinsames Merkmal der Erkrankung identifizieren.3 Demzufolge wird die LCH seit 2010 als entzündliche myeloische Neoplasie klassifiziert. In der Pathologie zeigt sich eine Akkumulation klonaler CD207-positiver Zellen der Monozyten-Makrophagen-/dendritischen Zelllinie. Sie proliferieren nicht, zeigen aber ein entzündliches Umgebungsinfiltrat aus Makrophagen, T-Zellen und neutrophilen Granulozyten. Dies führt zu einer lokalen Gewebsdestruktion, welche sich auch in lokalen oder systemisch erhöhten Inflammationsparametern widerspiegelt. Die breite Spanne der Krankheitsbilder wiederum hängt mit dem Entwicklungsgrad der Ursprungszelle zusammen. Dabei war eine relevante Erkenntnis der letzten Jahre, dass LCH-Läsionen eben nicht von den Langerhanszellen der Haut ausgehen, sondern von Zellen der myeloischen Differenzierung und damit von hämatopoetischen Stammzellen des Knochenmarks. Eine auf Stamm- oder Progenitorzellebene erworbene Mutation des MAPK-Kinase-Signalwegs führt daher zu einer Hochrisiko-Multisystem-LCH, während bei Mutationen eines DC-Progenitors oder eines Monozyten auf Gewebsebene eine Niedrigrisiko-Situation entsteht, in der aber ein Multisystembefall auftritt. Lokalisierte Verläufe hingegen treten dann auf, wenn lokale Präkursoren dendritischer Zellen betroffen sind. Eine Sonderstellung nimmt hier die neurodegenerative LCH ein, die infolge einer erfolgreich therapierten systemischen LCH auftreten kann oder als primäre zerebrale LCH ausgehend von fötalen Progenitoren. Zusammenfassend ist festzustellen, dassder Differenzierungsgrad der Ursprungszelle das klinische Bild entscheidet.
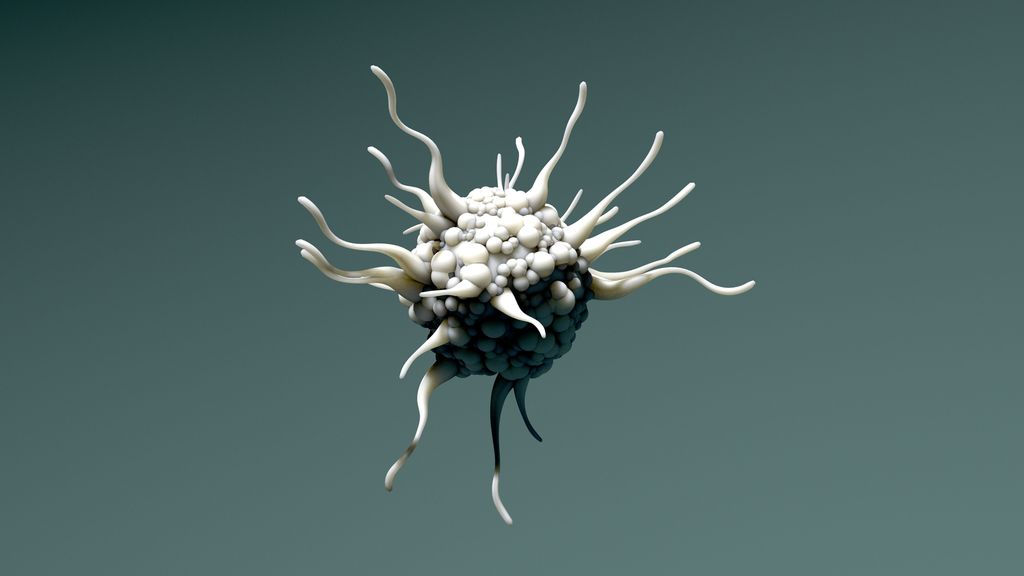
Untersuchungen des Mutationsspektrums zeigen in über 80% der LCH-Fälle eine aktivierende Mutation des MAPK-Signalwegs, vor allem die BRAFV600E-Mutation, gefolgt von MAP2K1-Mutationen. Letztere können zur «oncogen-induced senescence» führen, wie kürzlich an Arbeiten im Mausmodell gezeigt werden konnte.4 Dabei handelt es sich um ein Phänomen, bei dem Zellen einen Zell-Zyklus-Arrest in der Zellzyklusphase G1 sowie eine Apoptoseresistenz, morphologische Veränderungen und die Produktion eines inflammatorischen Sekretoms aufweisen. Diese Veränderungen erklären die Besonderheiten der LCH. Nicht eine Zellproliferation, sondern das inflammatorische Sekretom der LCH-Zellen ist verantwortlich für die lokale Gewebsdestruktion und Infiltration durch Zellen der adaptiven und angeborenen Immunabwehr (Abb. 1). Ob diese Veränderungen auch therapeutisch adressiert werden können, ist derzeit noch unklar. Präklinische Daten zeigen eine Wirksamkeit von das inflammatorische Sekretom modulierenden sogenannten Senomorphika wie mTOR-Inhibitoren oder Senolytika wie dem BCL-xl-Inhibitor Navitoclax, die zu einer Depletion seneszenter Zellen führen. Für Letzteres ist aktuell eine klinische Studie in Planung.
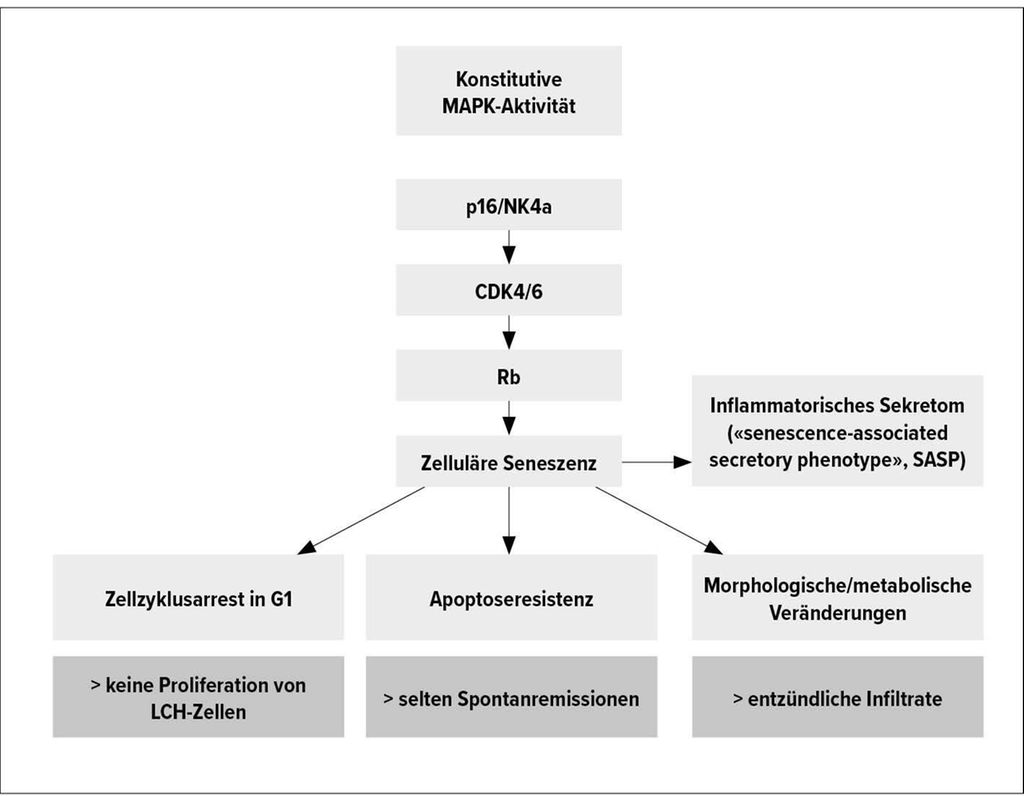
Abb. 1: Onkogen-induzierte Seneszenz (nach Wilk M, DGHO 2020)
Diagnostik und Therapie: Ein interdisziplinärer Ansatz ist notwendig
Die aktuellen therapeutischen Strategien wurden anschliessend von Prof. Dr. med. Christoph Klein (Ärztlicher Direktor der Kinderklinik und Kinderpoliklinik im Dr. von Haunerschen Kinderspital der Ludwig-Maximilians-Universität München), erläutert. Vor Beginn der Therapie erfolgt eine Risikostratifizierung der Erkrankung. Wesentlich ist hier nicht nur die Frage, ob es sich um einen isolierten Befall, die Single-System(SS-)LCH, handelt oder einen Mehrorganbefall (Multisystem[MS]-LCH). Ebenso muss beurteilt werden, ob ein Risikoorganbefall vorliegt (Knochenmark, Leber, Milz). Hieraus leiten sich die Therapiestrategien ab. Bei einem isolierten Befall des Knochens oder der Haut kann ein exspektatives Verfahren gewählt werden oder es kommen lokale Verfahren zum Tragen (Radiatio oder lokale Steroidapplikationen bei ossären Läsionen, Phototherapie bei kutanem Befall). Sind Risikoorgane betroffen, ist eine systemische Therapie notwendig. Im Kindesalter ist die Therapie mit Vinblastin und Prednison, 6-Mercaptopurin und Methotrexat ebenso wie der Einsatz von Nukleosidanaloga (Cytarabin, Cladribin, Clofarabin) gut belegt. Die Situation bei erwachsenen Patienten ist schwieriger. Die Kombination Vinblastin und Prednison wird deutlich schlechter vertragen. Daher werden hier in der ersten Linie Nukleosidanaloga bevorzugt eingesetzt. Mit der Erkenntnis der Bedeutung von MAPK-Signalwegs-Mutationen kommen neue Medikamente infrage, die eine gezielte Therapie ermöglichen. BRAF-Antagonisten (Vemurafenib, Dabrafenib) oder MEK-Inhibitoren (Cobimetinib, Trametinib) können hier direkt eingreifen. Aktuell werden diese Substanzen aber oft erst in der Rezidivsituation eingesetzt (Tab. 1).

Tab. 1: Empfehlungen zur Erstlinientherapie der LCH analog Eurohistionet
Dr. med. Claudia Lang, Oberärztin an der Klinik für Dermatologie und Allergologie am Universitätsspital Zürich, unterstrich in ihrem Beitrag die Wichtigkeit der interdisziplinären Betreuung der oft komplex erkrankten Patienten. Die Haut ist nach den Knochen das am zweithäufigsten betroffene Organ. Klinisch manifestiert sich dies typischerweise in Form eines seborrhoischen Exanthems sowie erythematöser Plaques vor allem an der Kopfhaut und in Körperfalten. Ein isolierter Hautbefall ist oftmals selbstlimitierend, bedarf aber eines engmaschigen Monitorings. Bei einer Behandlungsindikation variieren die Möglichkeiten zwischen dem Einsatz topischer Steroide oder Tacrolimus, Phototherapie oder auch der Exzision der Läsion. Bei ausgedehnterem Befall muss eine systemische Therapie in Betracht gezogen werden. Angesichts der oft störenden Symptome (Sichtbarkeit, Juckreiz) besteht eine Einschränkung der Lebensqualität und damit eine Therapieindikation. Zusätzlich kann der Hautbefall mit geringem Aufwand diagnostisch abgeklärt werden. Eine dermatologische Vorstellung sollte daher Teil jedes diagnostischen Work-ups bei histiozytären Erkrankungen sein.
Züricher Histiozytose-Register
Der interdisziplinäre Gedanke trägt auch die Idee des aktuell im Aufbau befindlichen Züricher Histiozytose-Registers am Universitätsspital Zürich, welches unter anderem Gegenstand des abschliessenden Vortrags «Langerhanszell-Histiozytose: Trials und Register im Erwachsenenalter» (Dr. med. Wiebke Rösler, Zürich) war. Angesichts der niedrigen Fallzahlen liegen kaum randomisierte Studien vor. Eine multizentrische, internationale Studie musste 2013 mangels Rekrutierung abgebrochen werden. Die aktuellen Therapieempfehlungen basieren daher im Wesentlichen auf retrospektiven Daten. Cantu zeigte 2012 Ergebnisse zur Wirksamkeit der Monotherapie mit Cladribin, Cytarabin und Etoposid anhand einer Analyse von 57 Patienten.52017 belegte Laird an Daten von 39 Patienten, die über einen Zeitraum von 20 Jahren bei isolierten ossären Läsionen mit einer Radiatio behandelt worden waren, das sehr gute Ansprechen dieser therapeutischen Massnahme.6 In Hinblick auf den Einsatz von BRAF-Inhibitoren ist der VE-Basket Trial (Diamond et al., 2019) die wesentlichste Studie (22 Patienten mit Erdheim-Chester-Erkrankung, 4 Patienten mit LCH). Gleichzeitig wird immer wieder eine hohe Dunkelziffer in der Diagnose bei Erwachsenen diskutiert. Auf Basis dieser Überlegungen sind bisher Kollegen der medizinischen Onkologie und Hämatologie, Endokrinologie, Dermatologie, Pathologie und Radiologie am Universitätsspital Zürich zusammengeschlossen, um sowohl die Diagnostik als auch die therapeutischen Optionen zu verbessern. Ferner soll mittels eines Registers eine solide Datenbasis erstellt werden, die einen genaueren Überblick über die Erkrankung gewährleisten und Basis klinischer Studien in der Zukunft darstellen soll. Neben dem Universitätsspital Zürich haben innerhalb der Schweiz bereits das Inselspital Bern, das Universitätsspital Basel, das Kantonsspital St.Gallen und das Ente Ospedaliero Cantonale Bellinzona ihre Mitarbeit signalisiert.
Literatur:
1 Carl E et al.: How I treat Langerhans cell histiocytosis. Blood2015; 126(1): 26-35 2 Baumgartner I et al.: Langerhans’-cell histiocytosis in adults. Med Pediatr Oncol 1997;28(1):9-14 3 Carl E et al.: Langerhanszell-histiocytosis. N Engl J Med 2018; 379:856-68 4 Anahita Rafiei et al.: BRAFV600E or mutant MAP2K1 human CD34+ cells establish Langerhans cell–like histiocytosis in immune-deficient mice. Blood Adv 2020; 4(19): 4912-7 5 Cantu MA et al.: Optimal therapy for adults with Langerhans cell histiocytosis bone lesions.PLoS One one2012; 7(8):e43257 6 Laird J et al.: Outcome after radiation therapy for langerhans cell histiocytosis is dependent on site of involvement. Int J Radiat Oncol Biol Phys 2018; 100(3): 670-8
Das könnte Sie auch interessieren:
Erfolg zielgerichteter und zeitlich begrenzter Therapien
Auf der Frühjahrstagung der OeGHO widmete sich eine spezielle Session zukunftsweisenden Entwicklungen in der Grundlagen- und klinischen Forschung auf dem Gebiet der Hämatologie. Im Fokus ...
Bessere Prognose durch MARS-R (Revised Mutation-Adjusted Risk Score)
Durch die Einführung moderner KIT-gerichteter Therapien ist die prognostische Aussagekraft bisheriger Risikomodelle eingeschränkt. Vor diesem Hintergrund wurde der MARS-R entwickelt.
Zelluläre Therapien ab der 2. Linie
Während die Erstlinienbehandlung des follikulären Lymphoms (noch) fest in der Hand der Chemoimmuntherapeutika liegt, sind bereits ab der zweiten Linie vermehrt zelluläre Therapien auf ...