
Gentherapeutische Ansätze zur Behandlung der Hämophilie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
In der Therapie der Hämophilie sind in den letzten Jahren viele Fortschritte erzielt worden. Aber trotz Halbwertszeit-verlängernder Präparate oder «Non-factor»-Präparate, die es ermöglichen, die ansonsten notwendige i.v. Applikation zu umgehen, kommt es im Rahmen der Therapie zu «Durchbruchsblutungen» und generell zu einer hohen Therapiebelastung. Die Gentherapie bietet erstmalig die Chance auf eine dauerhafte Heilung der Erkrankung. Diese Übersicht zeigt die erreichten Resultate, aufgetretenen Probleme und entstandenen Möglichkeiten.
Keypoints
-
Die Gentherapie stellt eine potenzielle Heilungsmethode der Hämophilie dar.
-
Mehrjährige Nachbeobachtungen nach Gentherapie zeigen eine gute Effizienz mit deutlicher Verminderung der jährlichen Blutungsraten und des Faktorverbrauchs, wobei aktuell die Dauer des Therapieeffekts unbekannt ist.
-
Die Weiterentwicklung des Vektordesigns ermöglicht noch höhere Effizienz und weniger Nebenwirkungen.
-
Eine lebenslange Nachbeobachtung von Patienten nach Gentherapie ist durch Aufbau eines weltweiten Registers durch die WFH geplant.
Die Hämophilie ist eine X-chromosomal vererbte monogene Erkrankung. Zudem entsteht sie in ca. 25% aus Neumutationen. Die Hämophilie A, die zu einer verminderten oder fehlenden Aktivität des Gerinnungsfaktors VIII führt, wird bei einem von 5000 männlichen Neugeborenen nachgewiesen. Die Hämophilie B, die den Faktor IX betrifft, ist seltener und tritt bei einem von 25000 männlichen Neugeborenen auf. Je nach Verminderung der Aktivität teilt man sie in eine schwere (<1% Restaktivität, Referenzbereich von 50–200%), moderate (1–5%) oder milde (>5% Restaktivität) Hämophilie ein.
Schwere und auch teilweise moderate Hämophilie sind durch spontane Gelenk- und Muskelblutungen charakterisiert. Die Behandlung besteht in einer prophylaktischen Verabreichung von Gerinnungsfaktoren durch die Patienten oder einer Bedarfsbehandlung im Blutungsfall. Obwohl die Prophylaxebehandlung mittlerweile als Standard für die Therapie der schwereren Hämophilie gilt, ist sie durch wiederholte i.v. Faktorsubstitution belastend. Ein Patient mit schwerer Hämophilie A muss so z.B. 150x/Jahr ein Standard-Halbwertszeit-Präparat injizieren. Durch die Modifikation der rekombinanten Gerinnungsfaktoren (Pegylierung, Fusion mit Fc-Teilen oder Albumin) konnte eine Verlängerung der Halbwertszeit um das 1,5-Fache bei Faktor-VIII-Präparaten und um das 5-Fache bei Faktor-IX-Präparaten erzielt werden. Je nach Blutungstyp und Präparat erfordert dies dennoch ca. 50–100 Injektionen, sodass die Compliance nicht immer gewährleistet ist. Zudem ist die Therapie durch die Entwicklung von «Anti drug»-Antikörpern, sogenannten Hemmkörpern, in bis zu 30% der Fälle bei Hämophilie A erschwert. Auch kommt es zu Durchbruchsblutungen und Hämophiliearthropathie, die von einer Verminderung der Lebensqualität bis zur Behinderung führt.1
Um diese Problematiken zu umgehen, wurden sogenannte «Non-factor»-Präparate entwickelt, die entweder Faktor VIII imitieren (wie z.B. Emicizumab) oder die das Blutungspotenzial des Patienten in Richtung «Thrombophilie» verschieben wie z.B. «small interfering» RNA zum «antithrombin silencing», Anti-TFPI(«Tissue factor pathway inhibitor»)-Antikörper oder die Inhibition von Protein C oder S.2
Potenzial der Gentherapie
Bei einer monogenen Erkrankung wie der Hämophilie bietet die Gentherapie die Möglichkeit, das fehlende oder nicht funktionierende Gen zu ersetzen. Hierbei ist ein effizienter Transfer in die zukünftig produzierenden Zellen notwendig. Um dies zu erreichen, können verschiedene Wege eingeschlagen werden, wie eine In-vivo-Genaddition mittels adeno-assoziierter Viren (AAV) oder ex vivo mittels Lentiviren zur «Korrektur» hämatopoetischer Stammzellen. Zukünftig ist auch der Einsatz von CRISPR(«Clustered regularly interspaced short palindromic repeats»)/Cas zur Geneditierung denkbar.3
Studien zur Gentherapie
Erste Gentherapieversuche vor knapp 20 Jahren führten nicht zu anhaltenden Faktorerhöhungen, sodass in den letzten Jahren in erster Linie rekombinante AAV verwendet wurden.4 Diese Viren sind nicht humanpathogen und tragen keine kodierenden Sequenzen der Wildtypen mehr. Sie sind durch einen «Lebertropismus» charakterisiert, sodass sie nach i.v. Infusion zielgerichtet Leberzellen transfizieren. Die typische Struktur eines rekombinanten AAV ist in Abb. 1 dargestellt. Sie besteht aus der gewünschten Gensequenz, die von Promotor- und Terminator-Sequenzen umgeben ist, wie auch an beiden Enden von «inverted terminal repeats» (ITR). Limitierend hierbei ist die maximale Gengrösse von 4,7kB, die für den Faktor IX mit einer Grösse von 2,8kB kein Problem darstellt, allerdings aber für den Faktor VIII mit einer Gengrösse von über 4,8kB. Deshalb verwendet man ein «B-domain»-verkürztes Faktor-VIII-Molekül, da die B-Domäne für die Aktivierung nicht notwendig ist.

Abb. 1: Struktur rekombinanter adenoassoziierter Viren (rAAV). ITR: «inverted terminal repeats»
Der rekombinante Genvektor wird als einmalige Infusion über 30 Minuten verabreicht. Nach Bindung des AAV-Genprodukts an glykosylierte Oberflächenrezeptoren der Leber werden die AAV-Partikel internalisiert, in Endosomen und Lysosomen prozessiert und anschliessend in den Nukleus transportiert (Abb. 2). Je nachdem, ob es sich um «selbstkomplementierende» AAV (sc-AAV), die direkt abgelesen werden können, oder um «Einzelstrang»-AAV (ss-AAV) handelt, müssen diese vor Transkription in Doppelstrang-DNA konvertiert werden. Das AAV bleibt episomal im Nukleus, wobei die ITR eine Ringform ermöglichen. Allerdings besteht die Gefahr, dass mit wiederholter Zellteilung ein «Verdünnungseffekt» auftreten kann, sodass dies unter Umständen im Falle einer wachsenden Leber wie im Kindesalter zum Funktionsverlust führen könnte. Neben der in erster Linie vorkommenden episomalen Persistenz der AAV konnte anhand von «deep sequencing» ein geringer Anteil von Integration ins Empfängergenom nachgewiesen werden. Allerdings konnte bisher keine Integration in die Nähe von «Driver»-Mutationen für hepatozelluläre Karzinome gefunden werden. In dieser Hinsicht wären dennoch zukünftige CRISPR/Cas-Ansätze mit gezielter Geneditierung vorteilhaft.5
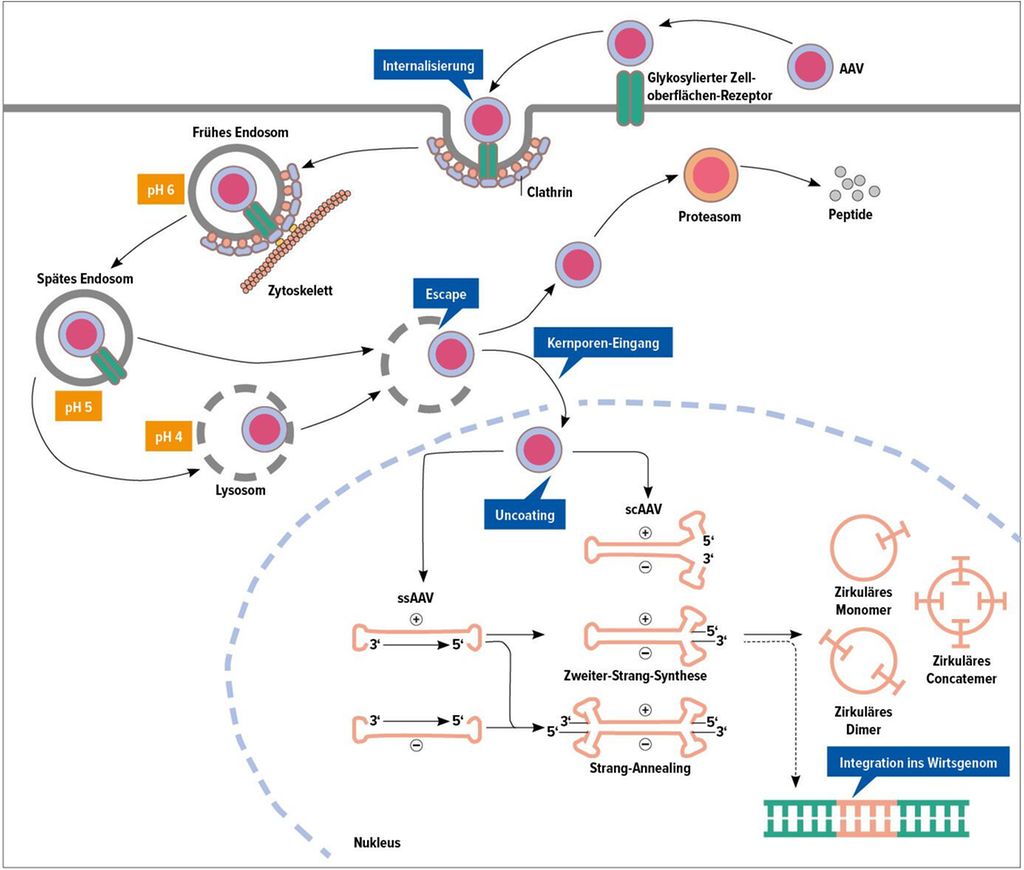
Abb. 2: Transduktionsmodus rekombinanter adenoassoziierter Viren (rAAV). Modifiziert nach Wang D et al.5
Die ersten erfolgreichen Gentherapiestudien mit anhaltender Persistenz der Faktorproduktion wurden 2011 bei Hämophilie B publiziert.6 Hierbei wurden verschiedene Vektordosen von 2x1011–2x1012vg/kg verwendet. In der Hochdosisgruppe kam es zu einem Anstieg auf ca. 5% Faktor-IX-Aktivität innerhalb der ersten 3 Jahre und einer Reduktion der Blutungsereignisse und des Faktorverbrauchs um 90%.7
Bei 4 der 6 Patienten dieser Gruppe kam es innerhalb der ersten 10 Wochen zu einer transienten Erhöhung der Alanin-Aminotransferase (ALT), die teilweise von einer Verringerung der Faktor-IX-Produktion begleitet war. Das Ausmass der Transaminasenerhöhung korrelierte mit der Vektordosis. Der Verlust der Faktor-IX-Produktion wie auch die Lebertoxizität wurden im Zusammenhang mit dem Auftreten von Kapsid-spezifischen zytotoxischen T-Zellen beobachtet.8 Eine orale Steroidtherapie führte unterschiedlich schnell zur Normalisierung der ALT. Zudem wurde eine fehlende Transduktion bei einem Patienten mit hohen, neutralisierenden Anti-AAV2-Antikörpern beobachtet. Dies führte dazu, dass man die AAV-Vektoren verbesserte, indem man Codon-optimierte Promotoren, «hyperfunktionelle» Faktor-IX-Varianten wie den «Faktor IX Padua» verwendete, der zu Faktor-IX-Aktivitäten von über 30% führte.9
Auch sc-AAV und weniger immunogene, unmethylierte Cytidin-Phosphat-Guanosin(CpG)-Motive wie auch andere Virus-Subtypen mit geringeren Inzidenzen von neutralisierenden Antikörpern (nAb) in der Bevölkerung wurden angewandt.
Trotzdem bleibt der Nachweis präexistenter nAb in den meisten bisherigen Studien ein Ausschlusskriterium, ebenso wie das Vorhandensein von Hemmkörpern bei Hämophilie A. 2019 gab es 12 aktive Trials in der Hämophilie B und 17 in der Hämophilie A. Trotz späteren Starts in der Hämophilie A ist es mittlerweile gelungen, auch hier verschiedenste Codon-optimierte Vektoren zu konstruieren,3 sodass die erste Zulassung für eine Gentherapie der Hämophilie A 2020 beantragt wurde. Diese wurde allerdings von der «Food and Drug Administration» (FDA) mit der Auflage einer längeren Nachbeobachtungszeit verschoben.10
Ausblick
In der Schweiz wurde innerhalb des Schweizerischen Hämophilie Netzwerkes (SHN), der nationalen Organisation der Hämophiliebehandelnden, eine Arbeitsgruppe «Gentherapie» gegründet, mit dem Ziel, Studienaktivitäten zu bündeln und den Patienten eine Teilnahme an ebensolchen zu ermöglichen. Berücksichtigt werden Ausschlusskriterien wie Alter <18 Jahren, vorbestehende nAb, Hemmkörper bei Hämophilie A, kardiovaskuläre Risikofaktoren, aber auch Sicherheitsaspekte wie Toxizität, Immunreaktion mit Transaminasenerhöhung, eventuelles Vektor-»Shedding» in andere Organe oder Integration in das Empfängergenom.
Die Effektivität dieser gentherapeutischen Ansätze ist sehr variabel, einerseits supranormale Faktoraktivitäten mit eventueller Thromboemboliegefahr oder andererseits Aktivitätsverlust. Ungeklärt ist bisher auch die Frage, ob eine erneute Verabreichung einer Gentherapie nach Entwicklung von Antikörpern gegen AAV möglich ist.
Die Erwartung der Patienten in Bezug auf Heilung ihrer Erkrankung mit einer Infusion ist gross und es bedarf sicherlich einer lebenslangen Nachbeobachtung wie von der World Federation of Hemophilia (WFH) initiiert.11
Literatur:
1 OʼHara S et al.: Disease burden and remaining unmet need in patients with haemophilia A treated with primary prophylaxis. Haemophilia 2020; online ahead of print 2 Weyand AC, Pipie SW: New therapies for haemophilia. Blood 2019; 133(5): 389-98 3 Batty P, Lillycrap D: Advances and challenges for haemophilia gene therapy. Human Mol Genetics 2019; 28(R1): R95-101 4 Gollomp KL et al.: Gene therapy for hemophilia: progress to date and challenges moving forward. Transfus Apher Sci 2019; 58(5): 602-12 5Wang D et al.: Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 2019; 18(5): 358-78 6 Nathwani AC et al.: Long-term safety and efficacy following systemic administration of self-complementary AAV vectorencoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 2011; 19(5): 876-85 7 Nathwani AC et al.: Long-term safety and efficacy of factor IX gene therapy in hemophilia. N Engl J Med 2014; 371(21): 1994-2004 8 Perrin GQ et al.: Update on clinical gene therapy for hemophilia. Blood 2019; 133(5): 407-14 9 George LA et al.: Hemophilia gene therapy with high specific-activity factor IX variant. N Engl J Med 2017; 377(23): 2215-27 10 Presseaussendung WFH: U.S. FDA rejects BioMarin hemophilia A gene therapy. 19. August, 2020 11 Konkle B et al.: Core data set on safety, efficacy, and durability of hemophilia gene therapy for global registry: communication from the SSC of the ISTH. J Thromb Haemost 2020; 18(11): 3074-7
Das könnte Sie auch interessieren:

Früherkennung klonaler Mastzellerkrankungen durch Screening im peripheren Blut
Die Diagnose einer systemischen Mastozytose und anderer klonaler Mastzellerkrankungen erfolgt in der klinischen Praxis häufig verzögert. Unspezifische und heterogene Symptome sowie die ...
Chronische myeloische Leukämie (CML)
Die Prognose der CML-Patient:innen unterscheidet sich durch die Entwicklung der Tyrosinkinaseinhibitoren (TKI) kaum mehr von der Normalbevölkerung. Dennoch gibt es neue Erkenntnisse, die ...
Therapieneuerungen: FL, CLL und LBCL
Am Jahreskongress der American Society of Hematology (ASH) wurden zahlreiche relevante Neuerungen in der Lymphomtherapie vorgestellt. Wir präsentieren drei ausgewählte Studien zur ...