Prä-, peri- und postoperatives Management neuroendokriner Tumoren
Neuroendokrine Tumoren haben eine Inzidenz von ca. 1–2/100000 Einwohner/Jahr und sind somit sehr selten, wenn auch die Inzidenz bei gewissen Tumorlokalisationen zunimmt. Zudem kommen neuroendokrine Tumoren im gesamten Gastrointestinaltrakt und sogar in der Lunge vor, da embryologisch die Lunge eine Ausstülpung des Gastrointestinaltrakts darstellt. Die Seltenheit und die Heterogenität mit sehr unterschiedlicher Biologie und folglich auch sehr unterschiedlicher Prognose bedingen, dass diese Tumoren meist sehr individuell behandelt werden. Zudem sind Studien wegen dergeringen Fallzahl schwierig und langwierig.
Keypoints
-
Die Chirurgie der NET sollte in jeder Erkrankungsphase in Betracht gezogen werden, da eine Zytoreduktion um 90% die weitere Behandlung mit Somatostatinanaloga und die Radiopeptidrezeptortherapie erleichtert. Zudem kann im Falle eines hormonellen Syndroms die hormonelle Symptomatik deutlich gebessert werden.
-
Bei metastasiertem NET sollten bei jeder Operation prä-, peri- und postoperativ Somatostatinanaloga verabreicht werden (SSA 100μg 2–3x/d s.c., 500μg in 500NaCl: 50μg/h i.v.), um eine lebensbedrohliche Karzinoidkrise zu vermeiden.
-
Der endokrine Chirurg sollte sich unbedingt intensiv mit der hormonellen Diagnostik neuroendokriner Tumoren befassen und in der Lage sein, die biochemische Diagnostik einzuschätzen. Bevor die biochemische Diagnose nicht klar ist, sollte keine Lokalisationsdiagnostik durchgeführt werden. Ein Beispiel wäre der Fall einer intrapankreatischen Nebenmilz, welche naturgemäß wie die „Hauptmilz“ sehr intensiv im Gallium-PET leuchtet und natürlich keinen Krankheitswert aufweist.
Neuroendokrine Tumoren (NET) sind entweder hormoninaktiv oder produzieren und sezernieren Substanzen, welche in gewissen Fällen ein hormonelles Syndrom verursachen. Somit besteht in gewissen Fällen für die Patienten eine Doppelerkrankung mit einem Tumor und einem hormonellen Syndrom, welches unbehandelt die Lebenserwartung in seltenen Fällen stärker reduzieren kann als die Tumorerkrankung selbst. So hat zum Beispiel das Cushing-Syndrom unbehandelt eine Mortalität von 50% innerhalb von 5 Jahren. Im Falle eines hormonellen Syndroms ist das Hormon selbst ein guter Tumormarker, sonst besteht mit Chromogranin A ein diagnostischerund therapeutischer Marker, welcher in 80–100% bei NET erhöht ist und sehr gut mit der Tumorload korreliert.
Bei NET mit ausgedehnter Lebermetastasierung kann in gewissen Fällen eine Fibrose der Pulmonal-und Trikuspidalklappe entstehen, was unbedingt einer präoperativen kardiologischen Beurteilung und evtentuell einer Klappenoperation bedarf. Seitdem mit den Somatostatinanaloga eine gute antisekretorische Therapie zur Verfügung steht, ist die Inzidenz der „Carcinoid Heart Disease“ deutlich zurückgegangen. Im Falle einer intraoperativen Manipulation bei der Operation eines neuroendokrinen Tumors, wie zum Beispiel einer Leberresektion bei neuroendokrinen Metastasen der Leber, kann es zu einer Einschwemmung von vasoaktiven Substanzen in den Kreislauf kommen mit resultierender Karzinoidkrise. Symptome der Karzinoidkrise sind Flushsymptomatik, Blutdruckabfall, Durchfall, Bronchokonstriktion und Hyperthermie. So eine Krise kann bei Narkoseeinleitung, Embolisierung, Beginn der Chemotherapie, bei Infektion oder Operation auftreten und ist lebensbedrohlich. Es ist deshalb empfohlen, bei ausgedehnten NET vor, während und nach der Operation Somatostatinanaloga zu verabreichen.
Insulinom
Insulinome sind immer intrapankreatisch gelegen, meistens kleiner als 2cm und in 90% benigne, deshalb ist die Enukleation die Therapie der Wahl. Ist ein Insulinom in der Nähe des Ductus pancreaticus lokalisiert, was leicht im Pankreascaudabereich vorkommen kann, empfiehlt sich eher die milzerhaltende Pankreaslinksresektion, um nicht eine folgenschwere Pankreasfistel zu riskieren. Bei allen endokrin-chirurgischen Erkrankungen ist es für den endokrinen Chirurgen essenziell, die biochemische Diagnose des hormonellen Syndroms zu verifizieren und schlussendlich zu akzeptieren. Diesbezüglich ist jedem Chirurgen, der einen organischen Hyperinsulinismus operiert, zu empfehlen, die Kriterien nach Service zu internalisieren. Gerade im Falle des Insulinoms ist dies von großer Bedeutung, da sonst unter Umständen eine stundenlange unnötige Exploration mit Rundumfreilegung des Pankreas und eventull unnötiger Resektion von Pankreasgewebe mit entsprechenden Komplikationsmöglichkeiten droht. Zu denken ist auch an das Münchhausen-Syndrom (Selbstinjektion von Insulin, Einnahme von Sulfonylharnstoffen) und an viele verschiedenste Läsionen wie kleine intrapankreatische Lipome, Hämangiome und im Caudabereich an intrapankreatische Nebenmilzen, welche in der Somatostatinrezeptorszintigrafie (Gallium-PET) sehr gut speichern. Insulinome haben im Gegensatz zu den Gastrinomen in der Regel eine niedrige Somatostatinrezeptordichte, deshalb ist es nicht zu empfehlen, die Hypoglykämien beim Insulinom mit Somatostatinanaloga zu behandeln, da damit eher die Glukagonsekretion gehemmt wird und somit die Hypoglykämien verstärkt werden.
Gastrinom
Beim Zollinger-Ellison-Syndrom sind die Tumorläsionen wegen der üblicherweise sehr guten Somatostatinrezeptorexpression mit dem Gallium-PET gut darstellbar, auch ist deshalb die antisekretorische Wirkung sehr gut, wobei mit PPI und Somatostatin die Patienten beschwerdefrei werden. Zu bedenken ist allerdings, dass 90% der Gastrinome maligne sind und metastasieren. 90% der Gastrinome finden sich im sog. Gastrinomdreieck (Cysticusmündung – unteres Duodenalknie – Übergang Pankreaskopf/Korpus). Das klassische Gastrinom ist ein 5mm-Tumor, häufig submukös im Duodenum lokalisiert. Bei der Operation eines Zollinger-Ellison-Syndroms empfiehlt es sich, intraoperativ eine Duodenoskopie durchzuführen und den OP zu verdunkeln, dann sieht man durch die Diaphanoskopie die submukös lokalisierten Gastrinome und kann dann direkt über dem Tumor duodenotomieren und das Gastrinom enukleieren.
MEN-I-Syndrom
Beim MEN-I-Syndrom treten in 40–70% ab dem vierten Lebensjahrzent multiple hormonaktive oder inaktive neuroendokrine Tumoren auf. Da die Tumoren sehr langsam progredient sind, wird die Thompson-Operation empfohlen. Sie besteht in einer Pankreaslinksresektion und Enukleation der Tumoren im Pankreaskopf (Abb. 1). Eine komplette Pankreatektomie würde das Problem des MEN-I-Pankreas definitiv lösen, allerdings würde der resultierende Brittle-Diabetes die Lebenserwartung unter Umständen stärker einschränken als die MEN-I-Tumorerkrankung.
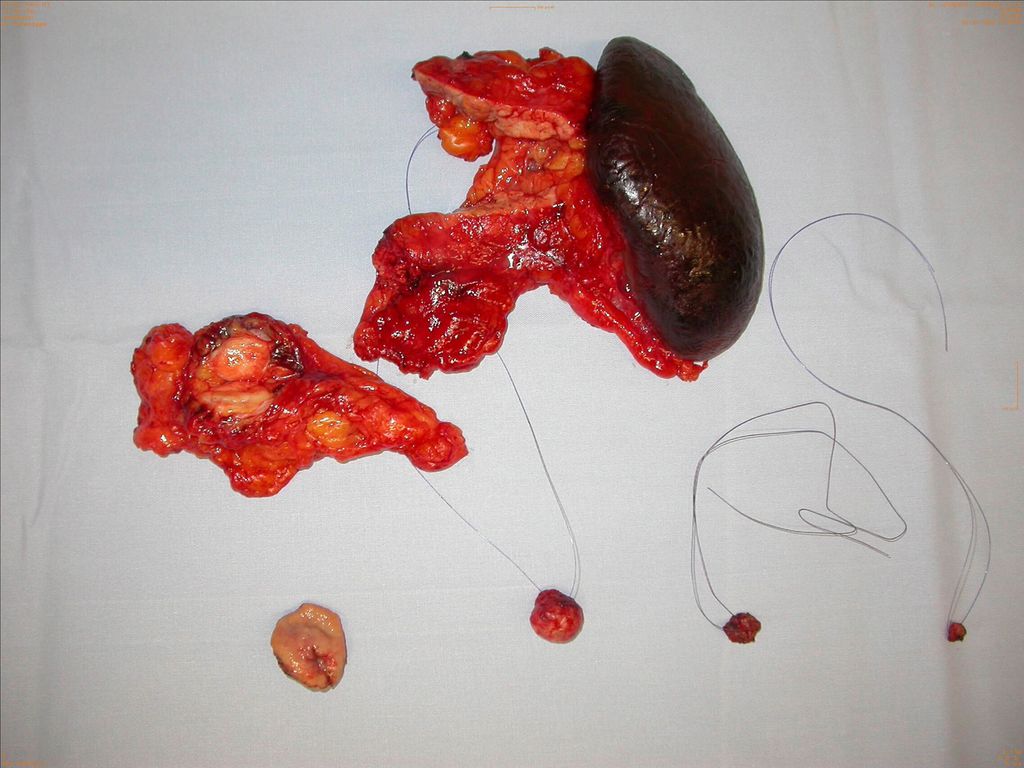
Abb. 1: OP-Präparate der sog. Thompson-Operation bei MEN-I-Syndrom
Pankreas-NET
Seltene hormonbildende Tumoren des Pankreas sind das Glukagonom, welches sich üblicherweise über ein stark juckendes Exanthem der Beine mit einem ausgeprägten Aminosäuremangel manifestiert. Präoperative parenterale Ernährung bessert den Gesundheitszustand schnell. Das VIPom ist meist ein relativ großer Tumor in der Pankreascauda. Die Symptomatik besteht in ausgeprägten wässrigen Diarrhöen, wobei literweise reine Elektrolytlösung abgesetzt wird. Mit Somatostatinanaloga sistiert die Diarrhö beim VIPom innerhalb von Stunden. Aufgrund der Vielfalt der neuroendokrinen Zellen können sich Tumoren im Pankreas entwickeln, welche bestimmte Neuropeptide sezernieren, die kein hormonelles Syndrom bewirken, allerdings kann das Neuropeptid als Tumormarker verwendet werden (z.B. NPY, PYY).
Dünndarm-NET
Neuroendokrine Tumoren des Dünndarms führen zu einer Induktion von Bindegewebe im Bereich des Dünndarmmesenteriums. Diese desmoide Reaktion findet sich um meist sehr große Lymphknotenmetastasen entlang des Dünndarmmesenteriums. Die desmoide Reaktion kann zu einer Durchblutungsstörung des Dünndarms mit entsprechender Symptomatik führen (Abb. 2). Diesbezüglich kann sich eine Indikation zur Notfalloperation ergeben, wobei der Chirurg wegen der desmoiden Reaktion mit einem großen „Tumorcake“ konfrontiert ist, der primär den Eindruck erweckt, es handle sich um einen Tumorbulk, welcher den gesamten Dünndarm einschließt. Dies führt gelegentlich durch die falsche Einschätzung zur inadäquaten Erstoperation. Meistens gelingt aber doch die Resektion unter Erhaltung eines ausreichend langen Darmsegmentes im oberen Jejunum. Der Primärtumor im Dünndarm ist meist sehr klein und führt zu keiner Obstruktion.
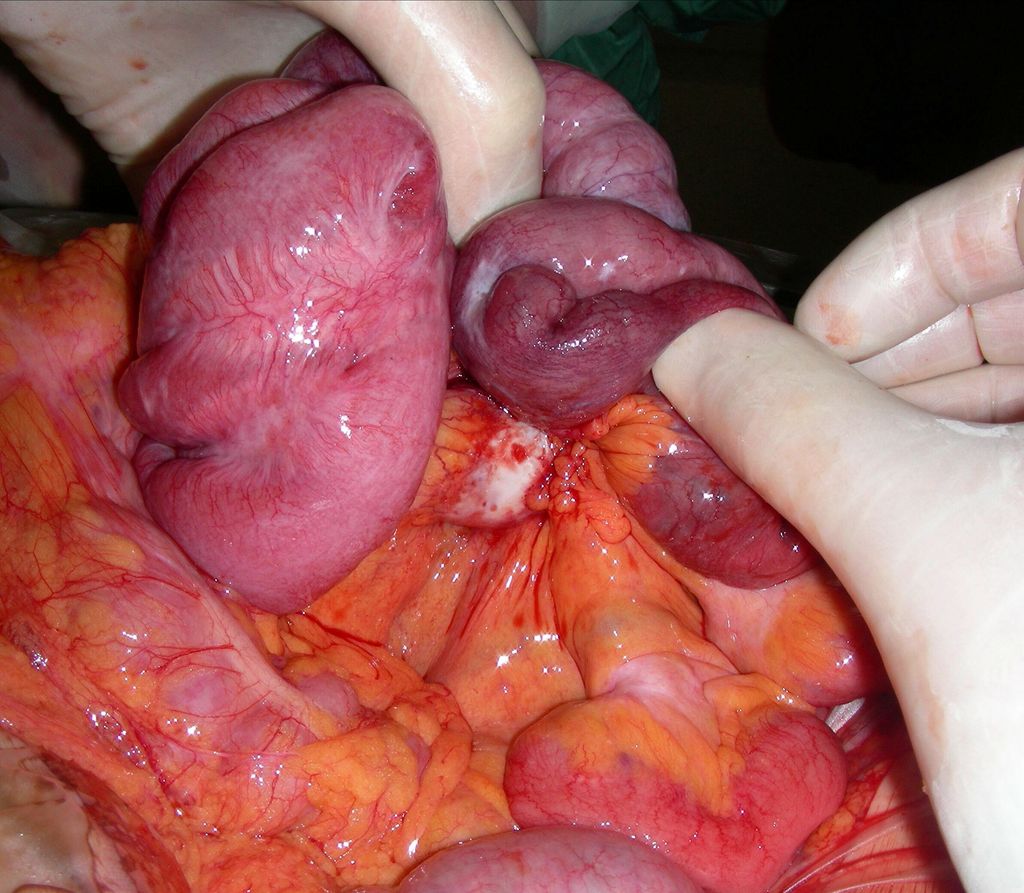
Abb. 2: NET des Dünndarms (Karzinoid) mit Lymphknotenmetastase am Mesenterialstiel (weiß), desmoider Reaktion und Durchblutungsstörung des Darms
Metastasierung
Bei den NET sollte trotz Metastasierung der Primärtumor entfernt werden, da dies das Langzeitüberleben verbessert. „Was bringt es, die Stalltür zuzuhauen, wenn das Pferd schon ausgebüxt ist.“ Bei den NET sollte man trotz Fernmetastasierung den Primärtumor entfernen, wenn das Operationsrisiko vertretbar ist. Eine Pankreaskopfresektion bei metastasiertem NET des Pankreaskopfes würde man wohl nicht durchführen. Bei jeder Operation wegen eines metastasierenden neuroendokrinen Tumors sollte die Gallenblase entfernt werden, da eine Therapie mit Somatostatinanaloga sehr wahrscheinlich früher oder später notwendig wird und diese die Gallensteinbildung fördern. Bei metastasiertem NET muss nicht notwendigerweise eine R0-Resektion erfolgen. Wenn 90% des Tumorvolumens mit vertretbarem Risiko entfernt werden können, ist dies günstig, indem z.B. ein hormonelles Syndrom wesentlich gebessert werden kann bzw. die Therapie mit „kaltem“ (Somatostatinanaloga) oder „heißem“ (Radiopeptidrezeptortherapie) Somatostatinanalogon effektiver wird.
Fazit
Aufgrund der Seltenheit und der Heterogenität der NET ist es für jeden Kollegen schwierig, die aktuellen Algorithmen zur Diagnostik und Therapie präsent zu haben. Diesbezüglich sei auf die Guidelines der ENETS (European Neuroendocrine Tumor Society) verwiesen, in der sich für jede spezielle Konstellation wohl eine brauchbare Empfehlung finden lässt!1
Literatur:
1 Pavel M, de Herder WW: ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors. Neuroendocrinology 2017; 105: 193-5
Das könnte Sie auch interessieren:

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

ASH 2020 – Highlights zu den aggressiven Lymphomen
Highlight-Themen der virtuellen ASH-Jahrestagung im Dezember 2020 waren an erster Stelle die Immunonkologika in all ihren Variationen, aber auch Beispiele für innovative Sequenztherapien ...

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...