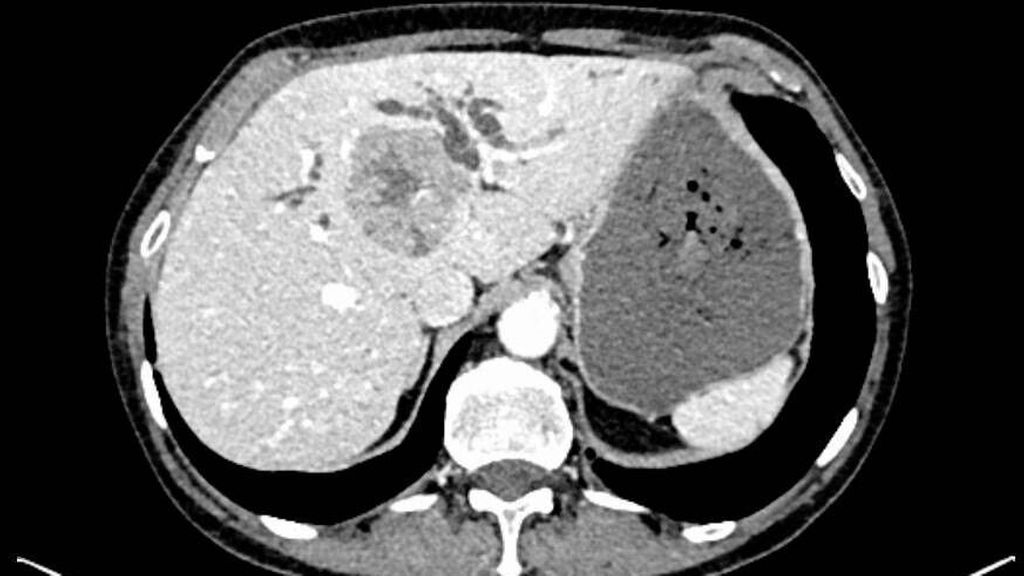
80-jährige Patientin mit kombiniertem hepatozellulärem Cholangiokarzinom
Im Oktober 2020 stellte sich eine 80-jährige Patientin erstmals in der GI-onkologischen Ambulanz Innsbruck vor. Sie berichtete über vor wenigen Wochen aufgetretene, von vermehrtem Aufstoßen begleitete, kolikartige Oberbauchschmerzen, die initial als Gastritis fehlgedeutet wurden. Seit einer Woche war sie zunehmend ikterisch. Bei der sonografischen Abklärung zeigte sich eine tumoröse Raumforderung der Leber.
Vorerkrankungen und Risikoprofil
Die Patientin war zum Zeitpunkt der Vorstellung 52kg schwer, 160cm groß (BMI: 20,5). Die Dauermedikation umfasste Seropram und Halcion. An relevanten Vorerkrankungen zeigten sich eine Post-HCV-Fibrose mit beginnender Zirrhose sowie mehrere kardiovaskuläre Risikofaktoren (arterielle Hypertonie, Hypercholesterinämie, prädiabetische Stoffwechsellage). Es bestand Zustand nach Cholezystektomie sowie Hysterektomie bei Frühkarzinom.
Klinische Befunde und Labor
Die Laboruntersuchung ergab ein deutlich erhöhtes Bilirubin (5,51mg/dl), leicht erhöhte Transaminasen (GOT: 86U/l, GPT: 55U/l) und erhöhte Cholestaseparameter (Gamma-GT: 287U/l, AP: 170U/l). Die Lebersyntheseleistung war erhalten (INR: 1,0; Thrombozyten: 241G/l; Albumin: 4,192mg/dl). Die Tumormarker waren auffällig:
-
AFP: 5,877µg/l
-
Ca19-9: 96U/ml
Bildgebung und Histologie
Die initiale CT zeigte eine bis zu 6cm große Raumforderung, ausgehend von der Hepatikusgabel, mit zentraler Infiltration des Leberparenchyms, suspekten Lymphknoten im Leberhilus und retroperitoneal, deutlich dilatierten intrahepatischen Gallengängen sowie Umscheidung und Infiltration des linken Pfortaderastes, jedoch ohne Thrombose. Der Befund war hochverdächtig auf einen cholangiozellulären Tumor. Die histologische Aufarbeitung einer Punktionsbiopsie ergab jedoch ein gering differenziertes, hiläres hepatozelluläres Karzinom (WHO 2019: G3). Angesichts des bildgebenden Befundes und des ebenfalls erhöhten Ca19-9 stand der Verdacht auf einen Mischtumor im Raum.
Therapie und Verlauf
Das interdisziplinäre Tumorboard bewertete den Tumor als inoperabel und empfahl eine Systemtherapie. Die Patientin erhielt zunächst zwei Zyklen Atezolizumab/Bevacizumab.
Darunter entwickelte sie nach wenigen Wochen eine wässrige, mehrmals täglich auftretende Diarrhö ohne Blutbeimengung, Übelkeit und Fieber bis 39°C. Die dritte Gabe erfolgte aufgrund der Beschwerden ohne Atezolizumab. In der weiterführenden endoskopischen Abklärung bestätigte sich eine immunvermittelte Kolitis. Die Patientin verlor 10kg Gewicht und musste stationär aufgenommen werden. Die immunvermittelte Kolitis wurde leitliniengerecht mit Methylprednisolon behandelt (1mg/kg, alle fünf Tage reduzierend über drei Wochen).
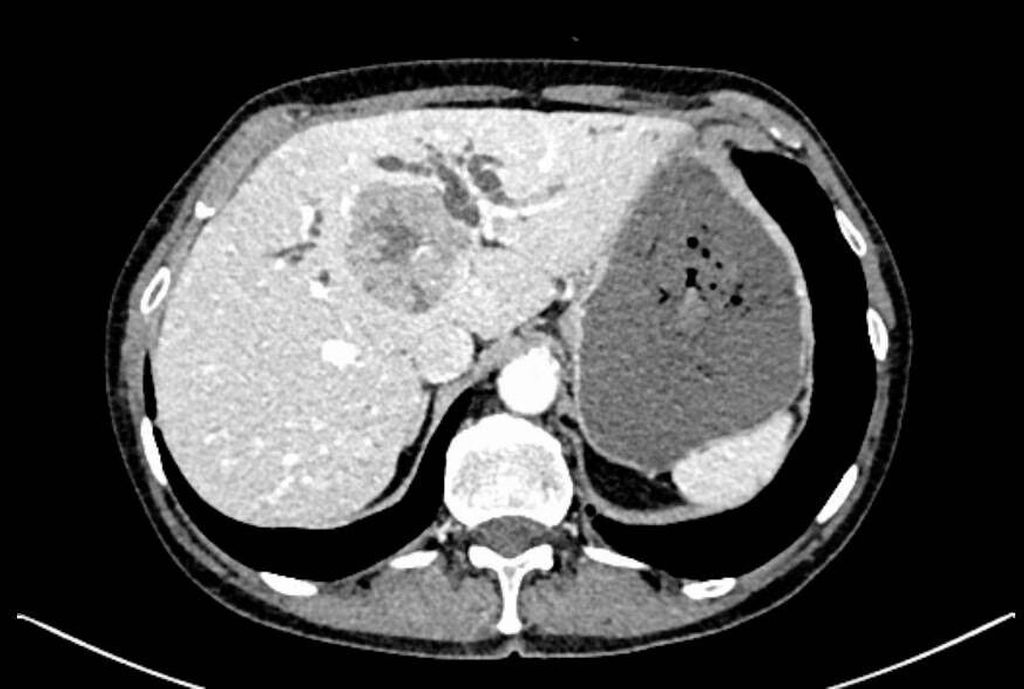
Abb. 1: CT bei Erstdiagnose
In der durchgeführten Staging-CT zeigte sich ein partielles Ansprechen (PR) mit Tumorgrößenreduktion von 49mm auf 33mm, die Lymphknoten blieben unverändert. Die Tumormarker sanken (AFP: 150, Ca19-9: 65). Aufgrund der persistierenden Diarrhö und des Verdachts auf einen Mischtumor erfolgte eine Umstellung der Therapie auf Cisplatin/Gemcitabin/Bevacizumab, wobei Gemcitabin ab Zyklus 2 wegen Fatigue auf 75% reduziert wurde und Cisplatin ab Zyklus 3 aufgrund von Ototoxizität abgesetzt werden musste. Nach vier Zyklen zeigte die MRT ein weiteres partielles Ansprechen (19×21mm), die Lymphknoten waren unverändert, die Tumormarker normalisierten sich erstmals.
Es erfolgte eine neuerliche Vorstellung im Tumorboard und es wurde die Entscheidung gefällt, eine Lokaltherapie durchzuführen. Im Juni 2021 erhielt die Patientin eine hypofraktionierte, extrakranielle stereotaktische Strahlentherapie (ED 12,5Gy, 3x, Zieldosis 37,5Gy auf die 65%-Isodose). Die Therapie wurde sehr gut vertragen. Im Verlauf zeigte sich ein anhaltendes komplettes Ansprechen (CR). Die Patientin befindet sich seither in regelmäßiger Nachsorge.
Im weiteren Verlauf traten rezente embolische, bihemisphärische Infarkte auf, ätiologisch als kardioembolisch bewertet. In der letzten MRT, viereinhalb Jahre nach Erstdiagnose, zeigten sich postradiogene Parenchymveränderungen, jedoch kein Hinweis auf ein Lokalrezidiv.
State of the Art:kombiniertes HCC-CCC
Definition und Epidemiologie
Das kombinierte hepatozelluläre Cholangiokarzinom (cHCC-CCA) ist eine seltene primäre Lebertumorentität, die Merkmale sowohl eines hepatozellulären Karzinoms (HCC) als auch eines intrahepatischen cholangiozellulären Karzinoms (iCCA) aufweist. Die WHO schätzt die Inzidenz auf 2–5% aller primären Lebertumoren. Die Erkrankung tritt durchschnittlich in einem Alter von 62–65 Jahren auf, Männer sind häufiger betroffen (Verhältnis circa 2:1).
Risikofaktoren und Pathogenese
Die Risikofaktoren für cHCC-CCA entsprechen denen der beiden Einzelkomponenten: Chronische Lebererkrankungen wie Hepatitis B und C, Leberzirrhose, Alkoholabusus und metabolisches Syndrom sind prädisponierend. In westlichen Ländern dominieren Hepatitis C und Alkohol als Ursache, in Asien Hepatitis B. Etwa die Hälfte der Patient:innen hat eine Leberzirrhose.
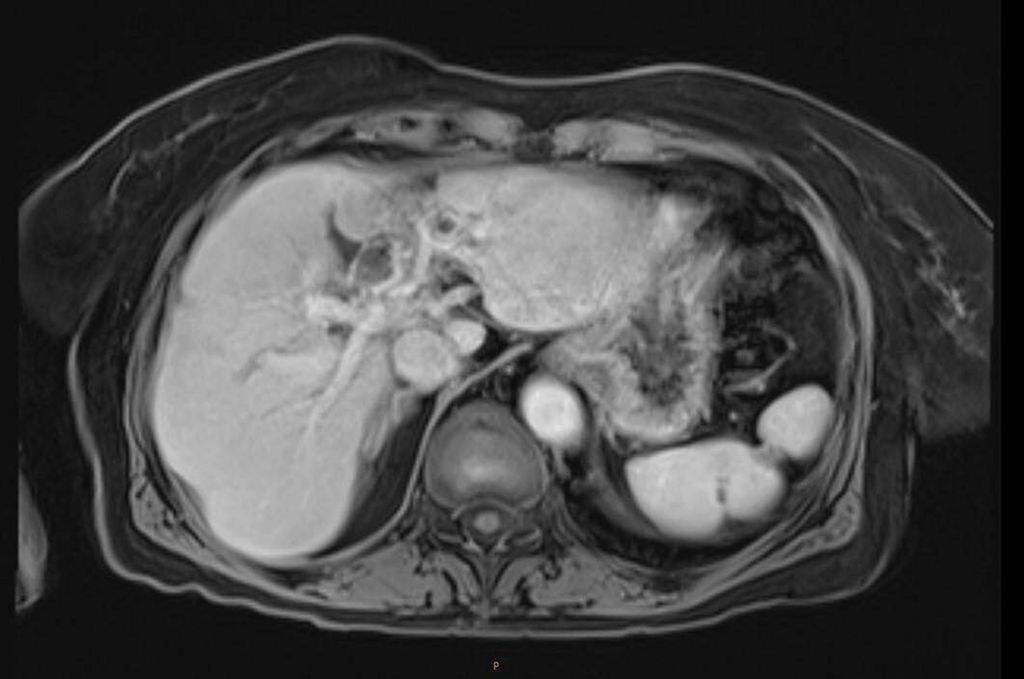
Abb. 2: MRT bei anhaltender kompletter Remission nach 4,5 Jahren
Die Pathogenese ist komplex und nicht abschließend geklärt. Molekulargenetisch zeigen cHCC-CCA-Tumoren eine hohe Heterogenität. Es wird angenommen, dass sie aus bipotenten hepatischen Vorläuferzellen hervorgehen können, die sowohl zu Hepatozyten als auch zu Cholangiozyten differenzieren können.
Histologie und Diagnostik
Die Diagnose setzt die eindeutige histologische Identifikation beider Differenzierungen voraus. Die aktuelle WHO-Definition (2019) verlangt das gleichzeitige Vorliegen von hepatozellulären und cholangiozellulären Tumoranteilen innerhalb eines Tumors. Immunhistochemisch werden Marker wie HepPar-1, Arginase-1, AFP (für HCC) und CK7, CK19, EMA (für iCCA) eingesetzt. Die Abgrenzung zu sogenannten „collision tumors“ (räumlich getrennte HCC- und iCCA-Herde) ist essenziell, da diese nicht als cHCC-CCA gelten.
Bildgebung
Bildgebend können sich Mischtumoren wie ein HCC-iCCA-Gemisch imponierend darstellen, wie ein typisches HCC oder aber ein typisches iCCA aussehen oder sich unspezifisch darstellen. Bildgebende Hinweise auf Mischtumoren bestehen bei verschiedenartigem Kontrastmittelverhalten innerhalb eines Tumors (im Verlauf zunehmende Anreicherung, Anreicherung mit Wash-out, Anreicherung ohne Wash-out oder Hypovaskularisierung), vaskulärer Invasion und Gallengangsbeziehungen. Die Kombination erhöhter Tumormarker (AFP und Ca19-9) kann einen Hinweis liefern, ist aber nicht beweisend.
Therapie
Kurative Therapieoptionen sind die chirurgische Resektion oder, in Einzelfällen, die Lebertransplantation. Allerdings sind bei Diagnose nur wenige Patient:innen operabel und die Rezidivrate ist hoch.
Aufgrund der Seltenheit der Erkrankung gibt es keine standardisierte Systemtherapie für inoperable Patient:innen. Die Behandlung orientiert sich mithilfe von Tumormarkern und Bildgebung an der dominierenden Tumorkomponente entsprechend der HCC- und iCCA-Leitlinien. Immuncheckpoint-Inhibitoren, VEGF-Hemmer, Tyrosinkinaseinhibitoren sowie Chemotherapien nach iCCA-Schema (z.B. Cisplatin/Gemcitabin) kommen zum Einsatz. Lokale Therapien wie TACE oder Radioembolisation können individuell erwogen werden. Um dem cholangiozellulären Anteil gerecht zu werden, sollte bei V.a. Mischtumor eine molekulare Aufarbeitung erfolgen, um Patient:innen eventuell eine zielgerichtete Therapie im Verlauf anbieten zu können.
Prognose
Die Prognose von cHCC-CCA ist schlechter als die von HCC und vergleichbar oder etwas besser als die von iCCA. Die Zeit des medianen Überlebens liegt unter zwei Jahren, mit großen Differenzen abhängig von Tumorstadium und Therapieansprechen. Molekulare Subklassifikationen und Biomarker könnten zukünftig helfen, gezieltere Therapiealgorithmen zu entwickeln.
Fazit
Der vorliegende Fall illustriert die Herausforderungen in Diagnostik und Therapie des cHCC-CCA. Die Patientin profitierte von Immuncheckpoint- und VEGF-gerichteter Therapie, Chemotherapie und stereotaktischer Strahlentherapie, was zu einer anhaltenden Remission führte.
Der Fall unterstreicht die Notwendigkeit weiterer Forschung und die Bedeutung interdisziplinärer Fallbesprechungen bei dieser seltenen, komplexen Tumorentität.
Literatur:
Ye L et al.: Combined hepatocellular-cholangiocarcinoma: biology, diagnosis, and management. Liver Cancer 2024; 13: 6-28
Das könnte Sie auch interessieren:

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

ASH 2020 – Highlights zu den aggressiven Lymphomen
Highlight-Themen der virtuellen ASH-Jahrestagung im Dezember 2020 waren an erster Stelle die Immunonkologika in all ihren Variationen, aber auch Beispiele für innovative Sequenztherapien ...

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...