
Hochheterogen und selten: das Mammasarkom
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Das Mammasarkom ist eine sehr seltene Erkrankung mit einer jährlichen Inzidenz von 4,6 Fällen pro einer Million Frauen. Unterschieden wird zwischen primären Formen und solchen, die z.B. im Zuge einer Strahlentherapie eines anderen Tumors entstehen können. Von den verschiedenen therapeutischen Optionen ist bislang die Resektion die einzige, welche potenziell kurativ ist. Die Behandlung des Sarkoms sollte an spezialisierten Zentren mit multidisziplinärer Betreuung der Patienten erfolgen.
Keypoints
-
Das Mammasarkom ist eine sehr seltene Erkrankung, bei welcher man zwischen primären und Therapie-induzierten Formen unterscheidet.
-
Der Schlüssel zur erfolgreichen Behandlung liegt in der Betreuung innerhalb spezialisierter, multidisziplinärer Sarkom- Boards.
-
Die am häufigsten auftretenden histologischen Subtypen im Bereich der Brust sind das Angiosarkom (vor allem Therapie-induziert), das Fibrosarkom und pleomorphe undifferenzierte Sarkome.
-
Die einzige kurative therapeutische Option ist die Resektion. Oberste Priorität hat dabei ein tumorfreier Resektionsrand.
Die prophylaktische Entfernung der Lymphknoten bei klinisch negativer Achselhöhle ist nicht indiziert.
Eine adjuvante Chemotherapie mit Doxorubicin und Ifosfamid wird eher bei Patientinnen mit Hochrisiko-Mammasarkomen empfohlen.
Bei weniger als 1% aller malignen Brusttumoren und weniger als 5% aller Weichteilsarkome handelt es sich um Mammasarkome. Die jährliche Inzidenz beläuft sich laut dem „Surveillance, Epidemiology, and End Results“(SEER)-Programm des National Cancer Institute auf 4,6 Fälle pro einer Million Frauen. Jedoch können auch Männer an Mammasarkomen erkranken.
Man unterscheidet zwischen primären und Therapie-induzierten (sekundären) Mammasarkomen, zu welchen vor allem das sekundäre Angiosarkom zählt. Über 95% aller sekundären Sarkome sind strahleninduziert. Aus histologischer Perspektive sind Sarkome hochheterogene, nicht epitheliale Tumoren, die aus dem Bindegewebe entstehen, mit über 100 bekannten Subtypen. Die in der Brust am häufigsten auftretenden Subtypen sind das Angiosarkom (Abb. 1), das Fibrosarkom und pleomorphe undifferenzierte Sarkome (PUS). Die Fibrosarkome und PUS liefern häufig das klinische Bild einer großen, festen, schmerzunempfindlichen Masse (nur selten mit Beteiligung der Haut), die schnell wächst. Die Diagnose erfolgt nicht selten überraschend im Zuge einer Operation, die der Entfernung eines eigentlich gutartigen Herdes dienen sollte. Angiosarkome hingegen gehen oftmals mit einer Verdickung der Haut, Erythemen oder einer Hautverfärbung ins Bläuliche einher. Therapie-induzierte Angiosarkome zeigen sich häufig durch kutane Läsionen, die entweder makulären oder papillären Erhebungen entsprechen.
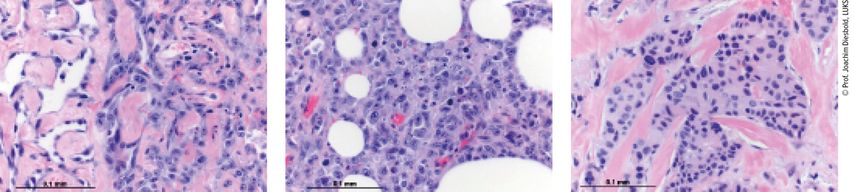
Abb. 1: Angiosarkome der Brust (links, Mitte) im Vergleich zum invasiven Mammakarzinom vom nicht speziellen Typ (NST; rechts)
Risikofaktoren, die zur Entstehung der Krankheit beitragen können, sind bei primären Mammasarkomen keine bekannt. Bei den sekundären Mammasarkomen wurde ein Zusammenhang mit ionisierender Strahlung im Zuge einer Strahlentherapie, mit bestimmten genetischen Prädispositionen (Li-Fraumeni-Syndrom, familiäre adenomatöse Polyposis [FAP], Neurofibromatose Typ 1 [NF1]) sowie mit chronischem Lymphödem berichtet. Ein sekundäres Mammasarkom gilt dann als durch Bestrahlung induziert, wenn drei Kriterien gegeben sind:
-
Ein maligner Tumor mit anderer Histologie als das durch Bestrahlung induzierte Sarkom wurde nachgewiesen;
-
das Sarkom entsteht an einer Stelle, welche im Rahmen der Strahlentherapie im Strahlenfeld gelegen ist;
-
die Latenzzeit zwischen dem Auftreten des ersten und des folgenden malignen Tumors ist recht lang (typischerweise mehr als vier Jahre).
Therapeutische Optionen
Resektion
Die einzige potenziell kurative Option in der Behandlung der Mammasarkome ist die Tumorresektion. Eine sorgfältige Planung ist dringlich zu empfehlen und sollte, wenn nötig, orthopädische und plastische Chirurgen sowie eine entsprechende Bildgebung miteinbeziehen. Die Resektionsränder sicher tumorfrei zu halten ist von größerer Wichtigkeit als die absolute Grösße der Ränder und hat einen direkten Einfluss auf das Langzeitüberleben der Patientinnen. Prinzipiell sollte ein Rand von einem Zentimeter eingeplant werden. Es sollte nicht davor zurückgeschreckt werden, Muskelgewebe zu entfernen, um größere basale Ränder zu erzielen. Außerdem ist zu beachten, dass insbesondere beim Angiosarkom multifokale und klinisch unauffällige Läsionen möglich sind.
Eine Analyse mittels Magnetresonanztomografie (MRT) kann die Abschätzung des Ausmaßes des Sarkoms erleichtern. Kapsulierte Tumoren sind möglich, besonders beim Fibrosarkom, dennoch wird die Entfernung der Kapsel allein nicht als ausreichend betrachtet. Ob eine Brusterhaltung sinnvoll ist, ist Gegenstand einiger Metaanalysen: Grundsätzlich sind Lokalrezidive sowohl nach Brusterhalt wie auch nach Mastektomie häufig. Die geforderten weiten Resektionsränder ziehen in der Praxis häufig zwangsläufig eine Mastektomie nach sich.
Mammasarkome metastasieren für gewöhnlich hämatogen. Nur selten sind benachbarte Lymphknoten (<5%) betroffen, weshalb die prophylaktische Entfernung bei klinisch negativer Achselhöhle nicht indiziert ist. Nichtsdestoweniger wurde im Rahmen einer Studie festgestellt, dass in den Vereinigten Staaten und in Europa ca. 40% der Patienten mit primärem Brustsarkom einer regionalen Lymphadenektomie unterzogen worden waren. Dieser Praxis entgegenzuwirken könnte dazu beitragen, die Morbidität im Zuge der Tumorbehandlung zu reduzieren. Klinisch suspekte Lymphknoten sollten vor einer großen Lymphadenektomie biopsiert werden.
Radiotherapie
Grundsätzlich ist kein Einfluss der Radiotherapie auf das Gesamtüberleben von Mammasarkompatientinnen festzustellen, es besteht nur die Möglichkeit, eine verbesserte lokale Kontrolle zu erzielen. Jedoch sollte stets ein Bewusstsein darüber bestehen, dass eine Radiotherapie keine Kompensation für eine mangelhafte Resektion darstellt.
Prinzipiell ist die Sinnhaftigkeit der Radiotherapie abhängig von Tumorgröße, Grading und Infiltration der umgebenden Strukturen. Speziell wird sie bei Mammasarkomen >5cm und Patientinnen mit positiven Resektionsrändern, die nicht weiter resektabel sind, empfohlen.
Bei großen, tiefgehenden Tumoren, bei denen mit positiven Rändern zu rechnen ist, kann eine präoperative Radiotherapie in Betracht gezogen werden, um die Resektabilität zu erhöhen. Bei strahleninduzierten Angiosarkomen kann im Einzelfall eine erneute Strahlentherapie angeboten werden.
Chemotherapie
Die Rolle der adjuvanten Chemotherapie beim Mammasarkom ist nicht klar definiert, die Daten werden aus Studien zu Weichteilsarkomen extrapoliert und die Zahl der tatsächlich behandelten Brustsarkome ist sehr unterschiedlich. Es gibt kaum Daten, die sich spezifisch der adjuvanten Chemotherapie beim Mammasarkom widmen. Dementsprechend schwierig ist das Aussprechen klarer Empfehlungen.
Eine Analyse der „Sarcoma Meta-Analysis Collaboration“ (SMAC) ergab moderate Überlebensvorteile für Patientinnen, die eine adjuvante Chemotherapie aus Doxorubicin und Ifosfamid erhielten, im Vergleich zur Resektion allein.
Generell wird eine adjuvante Chemotherapie mit einer Kombination aus Doxorubicin und Ifosfamid eher Patientinnen mit Hochrisikotumoren (nach Histologie, Größe und Grading) empfohlen. Andere Faktoren, die in die Entscheidung miteinbezogen werden sollten, sind der Performance Status, Komorbiditäten (inkl. Alter) und die Tumorlokalisation. Besonders bei Therapie-induzierten Sarkomen muss von Fall zu Fall eine fundierte Entscheidung unter Abwägung potenzieller Risiken getroffen werden.
Als weiteres potenzielles Chemotherapeutikum zeigt Paclitaxel bei Patientinnen mit Angiosarkomen im Stadium IV eine gute Wirksamkeit.
Zielgerichtete Therapien
Es wird angenommen, dass die Hochregulation angiogener Wachstumsfaktoren bei der Krankheitsentwicklung des Angiosarkoms eine wichtige Rolle spielt. In einer retrospektiven Analyse untersuchten Kollár et al. den Multi-Tyrosinkinase-Inhibitor Pazopanib beim Angiosarkom. Eine Krankheitskontroll-Rate von 40% sowie ein medianes progressionsfreies Überleben von drei Monaten konnten festgestellt werden.
Die Suche nach neuen therapeutischen Targets ist Gegenstand intensiver Forschung.
Nachuntersuchung
Rezidive entstehen häufig lokal und erfolgen typischerweise innerhalb der ersten drei Jahre. Fernmetastasierungen finden ebenfalls früh (<3 Jahre) statt und die Lunge ist häufig betroffen: Rezidive können in vielen Fällen erneut chirurgisch angegangen werden und viele Fälle mit Langzeitüberleben sind beschrieben.
Die „National Comprehensive Cancer Network“(NCCN)-Richtlinien empfehlen folgendes Vorgehen im Zuge der Nachuntersuchung: Patienten mit Mammasarkomen vom Grad II oder III sollten nach Therapieerhalt die ersten zwei bis drei Jahre alle drei bis sechs Monate (bildgebende Verfahren, physische Untersuchung) kontrolliert werden, danach für zwei Jahre alle sechs Monate. Nach Beendigung der Therapie eines Sarkoms vom Grad I wird die ersten zwei Jahre eine Nachuntersuchung alle drei bis sechs Monate, danach jährlich empfohlen. Die Grundidee der Nachsorge besteht in erster Linie darin, Rezidive in einem resektablen Stadium zu erkennen.
Prognose
Je nach individueller Situation der Patientin kann die Prognose stark variieren, vor allem abhängig von Histologie und Größe sowie Grading des Tumors. Nomogramme können zusätzlich hilfreich sein, um klinische Faktoren wie Alter und Tumorsitz miteinzubeziehen, was genauere Prognosen möglich macht.
Kattan et al. entwickelten ein Nomogramm zur Abschätzung der Wahrscheinlichkeit des Versterbens von Sarkompatienten in den ersten 12 Jahren nach einer Resektion. Diese einfachen Prognoserechner sind online verfügbar, z.B. unter
http://nomograms.mskcc.org/Sarcoma/PostSurgery.aspx.
Bericht:
Jasmin Gerstmayr, MSc
Review:
Prof. Dr. Peter Dubsky
Leiter BrustZentrum
Privatklinikgruppe Hirslanden
E-Mail: peter.dubsky@hirslanden.ch
Literatur:
Al-Benna S et al.: Diagnosis and management of primary breast sarcoma. Breast Cancer Res Treat 2010; 122(3): 619-26 • Casali PG et al.: Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010; 21(Suppl 5): v198-203 • Cugh R et al.: Breast sarcoma: Epidemiology, risk factors, clinical
presentation, diagnosis, and staging. UpToDate 2019.
Online unter https://www.uptodate.com/contents/breast-
sarcoma-epidemiology-risk-factors-clinical-presentation-
diagnosis-and-staging. Abgerufen am 4. Mai 2020 • Cugh R et al.: Breast sarcoma: Treatment. UpToDate 2019. Online unter https://www.uptodate.com/contents/breast-sarcoma-treatment. Abgerufen am 4. Mai 2020 • Kattan MW et al.: Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol 2002; 20(3): 791-6 • Kollár A et al.: Pazopanib in advanced vascular sarcomas: an EORTC Soft Tissue and Bone Sarcoma Group (STBSG) retrospective analysis. Acta Oncol 2017; 56(1): 88-92 •McGowan TS et al.: An analysis of 78 breast sarcoma patients without distant metastases at presentation. Int J Radiat Oncol Biol Phys 2000; 46(2): 383-90 • Pervaiz N et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008; 113(3): 573-81 • Ray-Coquard IL et al.: Paclitaxel given once per week with or without bevacizumab in patients with advanced angiosarcoma: a randomized phase II trial. J Clin Oncol 2015; 33(25): 2797-802 • Sheth GR et al.: Radiation-induced sarcoma of the breast: a systematic review. Oncologist 2012; 17(3): 405-18
Das könnte Sie auch interessieren:

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

ASH 2020 – Highlights zu den aggressiven Lymphomen
Highlight-Themen der virtuellen ASH-Jahrestagung im Dezember 2020 waren an erster Stelle die Immunonkologika in all ihren Variationen, aber auch Beispiele für innovative Sequenztherapien ...

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...