
„Alles steht und fällt mit der Diagnostik“
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Selten und hochkomplex zu therapieren: Sarkome werden in Österreich vor allem an spezialisierten Zentren behandelt. Wir sprachen mit Univ.-Prof. Dr. Thomas Brodowicz, österreichischer Experte auf diesem Gebiet, anlässlich seines Vortrags „Pathology – the basis of the sarcoma patients’ therapy“ bei der Frühjahrstagung der Österreichischen Gesellschaft für Pathologie über einige verschiedene Subentitäten, die pathologische und molekularpathologische Diagnostik sowie spezifische Therapieoptionen.
Wie ist die Sarkomtherapie in Österreich aufgestellt?
T. Brodowicz: Im letzten Jahrzehnt hat sich die Situation meiner Meinung nach durch die Zentralisierung stark verbessert. Sarkome werden jetzt hauptsächlich an Schwerpunktzentren diagnostiziert und behandelt. Als ich angefangen habe, war das noch nicht der Fall, da war das total verstreut. Mittlerweile ist es so, dass ich mehr als 50% der möglichen Sarkompatientenerstvorstellungen in Österreich sehe. Weitere Schwerpunktzentren gibt es u.a. in Graz und Linz. Ich möchte festhalten, dass die Kooperation der Internistischen Onkologen bezüglich Sarkomen sehr gut funktioniert. Sehr oft werde ich bezüglich Therapieempfehlungen kontaktiert (auch fachübergreifend), die ich in weiterer Folge sehr gerne gebe.
Wie hat sich die Therapielandschaft beim Sarkom in den letzten Jahren geändert?
T. Brodowicz: Einen wesentlichen Unterschied hat die Pathologie gemacht: Wir können und müssen nun ganz genau zwischen den mannigfaltigen Subtypen der Sarkome differenzieren. Dieser Fortschritt hatte natürlich Einfluss auf die Therapien, sodass nun für verschiedene Subentitäten spezielle Substanzen verfügbar sind. Daher hängt eine akkurate Therapie bei Weichteil- und Weichgewebssarkomen mit der pathologischen Diagnostik eng zusammen. Ich würde behaupten: Ohne genaue Histologie ist eine suffiziente Therapie nicht möglich. Die Zusammenarbeit mit der Pathologie ist deswegen von größter Bedeutung. Alles steht und fällt mit der Diagnostik. Wenn die Diagnostik nicht genau ist, hat das therapeutische Konsequenzen. Deswegen muss man die pathologischen Befunde auch kritisch lesen. Besonders wichtig sind hier die Tumorboards, um vor Ort gewisse Nuancen mit der Pathologie besprechen zu können. Das sind Details wie die Proliferationsrate der Zellen, ob mehr Knorpelsubstanz oder myxoide Substanz vorhanden ist oder ob es mehr Spindelzellen im Präparat gibt etc. Für mich alsInternistischen Onkologen ist es wichtig, diese Details zu kennen, um dann eine entsprechende systemische Therapie einleiten zu können.
Inwiefern ist die Erfahrung der Pathologie an Zentren ausschlaggebend?
T. Brodowicz: Es ist wesentlich, dass die Präparate von einer Pathologin begutachtet werden, die Erfahrung mit Sarkomen hat. Ich werde sehr oft für eine Zweit- oder Drittmeinung konsultiert. Mein erster Blick fällt dann immer darauf, wo das Präparat pathologisch begutachtet wurde. Wenn es sich dabei nicht um eine Referenzpathologie handelt, dann lasse ich mir als ersten Schritt die Paraffinblöcke organisieren und veranlasse die Begutachtung durch eine Referenzpathologin.
Wie setzt sich das Tumorboard an Ihrer Abteilung zusammen?
T. Brodowicz: Die wesentlichen Disziplinen bei der Diagnose und Behandlung von Patienten mit einem Sarkom sind (in alphabetischer Reihenfolge): Internistische Onkologie, Pathologie, Radiologie, Strahlentherapie, Tumororthopädie (z.B. bei abdominellen Sarkomen: Bauchchirurgie; bei thorakalen Sarkomen: Thoraxchirurgie). Das Tumorboard findet einmal pro Woche statt.
Wie läuft die Zusammenarbeit der unterschiedlichen Disziplinen ab?
T. Brodowicz: Sehr gut. Das Spektrum ist: Biopsieplanung, Besprechung der Histologie, Festlegung von Resektion, Strahlentherapie und systemischer Therapie plus Festlegung der Sequenz der Therapiemodalitäten und selektive Planung von Metastasenchirurgie und lokal-ablativen Verfahren. Das ist u.a. auch für die Pathologie sehr aufschlussreich. Die Verhältnisse können aufgrund der Bildgebung begutachtet werden und es lässt sich abschätzen, ob es sich eher um einen homogenen oder inhomogenen Tumor handelt. Die Pathologin weiß dann auch, wo bei der Biopsie die Nadelführung liegt. Das ist eine wertvolle Information, da Sarkome aus mehreren Anteilen bestehen können. In weiterer Folge ist es im Rahmen der Gewebsentnahme bei der Operation ganz wichtig, das Präparat in allen Dimensionen zu markieren. Die Pathologin muss sich orientieren können und muss auch den Resektionsrand bestimmen. Bei den Weichteilsarkomen ist der Resektionsrand der heikelste prognostische Parameter: Resektion durch sarkomerfahrene Chirurginnen ist essenziell.
Hat der Resektionsrand dann auch eine therapeutische Konsequenz?
T. Brodowicz: Korrekt. Beim Sarkom gibt es die Einteilung in marginale, weite und intraläsionale Resektion. Des Weiteren wird der Resektionsrand metrisch angegeben.
Die Sarkome sind eine sehr heterogene Gruppe von Malignomen. Im Verlauf Ihres Vortrags haben Sie einzelne Gruppen beleuchtet. Beginnen wir mit den Liposarkomen: Welches Vorgehen hat sich in der Praxis bei Ihnen bewährt?
T. Brodowicz: Bei den Liposarkomen gibt es mehrere Subtypen, z.B. das myxoide, dedifferenzierte und pleomorphe Liposarkom. In der metastasierten Situation wähle ich danach die Therapiesubstanzen aus. D.h., wenn es sich beispielsweise um ein myxoides Liposarkom handelt, dann wähle ich zuerst das Trabectedin. Wenn es sich hingegen um ein pleomorphes Liposarkom handelt, dann denke ich in erster Linie an das Eribulin, und wenn es ein dedifferenziertes Liposarkom ist, dann schaue ich mir an, wie der dedifferenzierte Anteil aussieht bzw. welcher histologischen Subentität er ähnelt, und wähle danach den Wirkstoff aus. Deswegen ist für mich die Aussage, dass es sich um ein Liposarkom handelt, zu wenig: Ich muss wissen, welcher Art es ist. Das ist nur durch erfahrene Pathologinnen festzustellen, u.a. weil auch die Molekularpathologie mit ins Spiel kommt. Die Diagnostik beruht also auf einer Kombination aus Histologie, Morphologie, Immunhistochemie und Molekularpathologie.
Beruht dieses Vorgehen auf festgelegten Leitlinien oder steht Ihre klinische Erfahrung dahinter?
T. Brodowicz: Es gibt Leitlinien. Allerdings bin ich davon überzeugt, dass die klinische Expertise bei Sarkomen nicht minder (wenn nicht sogar mehr) zählt. Das setzt natürlich eine entsprechende Patientenfrequenz voraus.
Als Nächstes kamen Sie auf das Synovialsarkom zu sprechen. Worin liegen häufige Probleme bei der Erstlinientherapie dieser Tumorentität?
T. Brodowicz: Das ist eine Entität, die vor allem jüngere Patienten betrifft. Auch hier ist in der Diagnostik die Kombination von Histologie und Molekularpathologie wichtig, denn das Synovialsarkom hat eine spezifische Genfusion. Es ist bekannt, dass das Synovialsarkom vor allem in der metastasierten Situation von einer suffizienten Dosis von Ifosfamid profitiert. Dabei handelt es sich um eine sehr hohe Dosis. Wenn man Patienten konsultiert, reicht es oft, die bisherige Ifosfamid-Dosis zu überprüfen – falls diese zu niedrig war, ist häufig die Therapie mit einer ordentlichen Dosis Ifosfamid ausreichend.
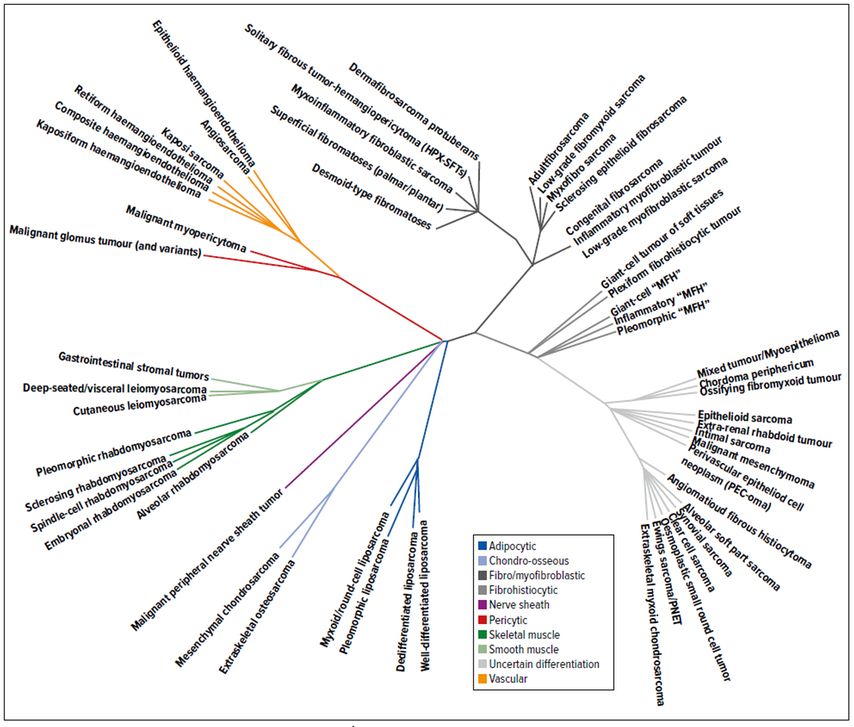
Abb. 1: Subentitäten der Sarkome (adaptiert nach Taylor et al.)1
Wie sieht es hier mit der Zweitlinie aus?
T. Brodowicz: In der Zweitlinie (auch Erstlinie oder „Mehrt“-Linie) muss man individuell entscheiden. Systemische Therapie, Metastasenchirurgie, Strahlentherapie oder lokal-ablative Therapien.
Worin liegen die Unterschiede zwischen dem uterinen und nicht uterinen Leiomyosarkom? Wie wirkt sich das auf das therapeutische Vorgehen aus?
T. Brodowicz: Es ist sehr wichtig, zwischen dem uterinen und nicht uterinen Leiomyosarkom zu unterscheiden. Die uterinen Leiomyosarkome sind klarerweise in der Gebärmutter, und die nicht uterinen finden sich vor allem an den Extremitäten, können aber z.B. auch im Thoraxraum oder im Bauchraum auftreten. Für mich sind die uterinen Leiomyosarkome prognostisch schlechter, das ist auch in der Literatur so beschrieben. Woran liegt das? Erstens sind sie bei Erstdiagnose viel größer als jene an den Extremitäten, da im Becken mehr Platz ist und sie daher erst relativ spät bemerkt werden. Zweitens sind sie oft bei Erstdiagnose metastasiert und das Metastasierungsmuster ist komplett anders als bei einem Extremitätenleiomyosarkom. Extremitätenleiomyosarkome metastasieren zumeist in die Lunge. Im Gegensatz dazu metastasiert das uterine Leiomyosarkom vor allem in das Bauchfell und in die Leber. Da Metastasen im Bauch sehr symptombehaftet sind, ist es wichtig, ein rasches Ansprechen auf die Therapie zu erzielen, um damit u.a. die Schmerzen zu lindern. Da die Patienten mit nicht uterinen metastasierten Leiomyosarkomen zumeist komplett asymptomatisch sind, ist in diesem Fall jedoch eine lange Stabilisierung komplett ausreichend. Deswegen sind das für mich zwei unterschiedliche therapeutische Ansätze.
Sie erwähnten auch, dass solitäre fibröse Tumoren (SFT) nicht gut auf die „klassischen“ Sarkomtherapien ansprechen. Welche Strategie verfolgen Sie bei dieser Erkrankung?
T. Brodowicz: Das ist eine Entität, die nur bedingt für die klassischen, systemischen Therapien sensibel ist. Vom biologischen Verlauf her gibt es zwei Subtypen: Den einen Subtyp können wir mit einer Operation heilen. Der zweite, aggressivere Subtyp metastasiert jedoch. Das Metastasierungsbild ist in diesem Fall etwas anders als bei den anderen Histologien, weil der SFT oft an den Hirnhäuten beginnt. Nicht selten wird er initial als Meningeom oder Ähnliches missklassifiziert. Jahre später kommt es dann zu einer singulären Lebermetastase oder einer Lungenmetastase, die fälschlicherweise als SFT der Leber bzw. Lunge klassifiziert wird, obwohl das Primum Jahre zuvor an den Hirnhäuten aufgetreten ist. Das ist ein Tumor, den ich schwer berechenbar finde. In der metastasierten Situation gebe ich z.B. eine Kombination von systemischer Chemo und Antiangiogenese. Auch hier ist es suffizient, die Krankheit über lange Zeit zu stabilisieren, man muss in vielen Fällen nicht unbedingt ein Ansprechen erzielen.
Beim SFT kann die Anamnese also wegweisend sein …
T. Brodowicz: Genau, in den Fällen von SFT in Leber oder Lunge führt man eine gründliche Anamnese durch und fragt den Patienten, ob er einmal eine Operation am Gehirn oder am Schädel gehabt hat. Dann weiß man sofort, was Sache ist.
Welche Rolle spielt die Molekularpathologie bei gastrointestinalen Stromatumoren (GIST)?
T. Brodowicz: Hier muss ich wissen, was für eine Mutation im konkreten Fall vorliegt, da das therapeutische Konsequenzen hat. Die Patienten mit einer Exon-9-Mutation benötigen in der metastasierten Situation die doppelte Dosis von Imatinib im Vergleich zu Patienten mit einer Exon-11-Mutation. Es gibt auch seltene Mutationen, wie die D842V-Mutation, die resistent auf nahezu alle Tyrosinkinaseinhibitoren sind. Seit ein paar Monaten ist in Europa nun das Avapritinib für metastasierte GIST mit dieser Mutation zugelassen. Hier kommt man also um die Molekularpathologie nicht herum, denn sie hat eine therapeutische Relevanz und Konsequenz.
Zum Abschluss besprachen Sie die Relevanz der Molekularpathologie beim Ewing-Sarkom.
T. Brodowicz: Die Ewing-Sarkome zählen zu den klein-, blau-, rundzelligen Sarkomen. Ohne Molekularpathologie könnte es sich aber auch um ein Synovialsarkom, ein alveoläres Rhabdomyosarkom, ein kleinzelliges Lungenkarzinom oder auch ein Lymphom handeln. Um das eindeutig feststellen zu können, benötigen wir die Molekularpathologie. Umso mehr, als es bei den Knochensarkomen gewisse Mutationen gibt, die prognostisch besonders schlecht sind. Wenn in einer Studie zum Ewing-Sarkom als Einschlusskriterium keine Molekularpathologie vonnöten war, stellt sich für mich die Frage, wie viele von den sogenannten Ewing-Sarkomen dann eigentlich gar keine waren oder eine prognostisch schlechtere Mutation aufwiesen. Das sind nicht unwichtige Nuancen.
Haben die Substanzen, die Sie verwenden, eine Zulassung für die einzelnen Entitäten?
T. Brodowicz: Wir reden hier von „orphan disease“. Und hierbei noch von mannigfaltigsten Subgruppen. Der Off-Label-Use ist für mich unverzichtbar, denn sonst könnte ich Sarkompatienten nicht ordentlich behandeln.
Wie würden Sie die Take-Home-Message Ihres Vortrags formulieren?
T. Brodowicz: Die ist relativ simpel: Wenn man ein Sarkom ordentlich behandeln will, muss man mit der Pathologin bzw. dem Pathologen sprechen.
Literatur:
1Taylor BS et al.: Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer 2011; 11(8): 541-57
Unser Gesprächspartner:
Univ.-Prof. Dr. Thomas Brodowicz
Programmdirektion Sarkome
Universitätsklinik für Innere Medizin
Klinische Abteilung für Onkologie
Universitätsklinikum AKH Wien
Medizinische Universität Wien
Das könnte Sie auch interessieren:

Highlights zu Lymphomen
Assoc.Prof. Dr. Thomas Melchardt, PhD zu diesjährigen Highlights des ASCO und EHA im Bereich der Lymphome, darunter die Ergebnisse der Studien SHINE und ECHELON-1

ASH 2020 – Highlights zu den aggressiven Lymphomen
Highlight-Themen der virtuellen ASH-Jahrestagung im Dezember 2020 waren an erster Stelle die Immunonkologika in all ihren Variationen, aber auch Beispiele für innovative Sequenztherapien ...

Aktualisierte Ergebnisse für Blinatumomab bei neu diagnostizierten Patienten
Die Ergebnisse der D-ALBA-Studie bestätigen die Chemotherapie-freie Induktions- und Konsolidierungsstrategie bei erwachsenen Patienten mit Ph+ ALL. Mit einer 3-jährigen ...