Hyperinflammation: Amoklauf des Immunsystems
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Eine ganze Reihe von Auslösern kann zu Hyperinflammationssyndromen führen, deren Gemeinsamkeit Zytokinsturm und potenziell resultierende Organschädigungen sind. Die hämophagozytische Lymphohistiozytose (HLH) und das VEXAS-Syndrom stellen zwei besondere Entitäten innerhalb dieser Gruppe dar. In beiden Indikationen werden mittlerweile Biologika und JAK-Inhibitoren erfolgreich eingesetzt.
Der Begriff „Hyperinflammationssyndrom“ ist nicht eindeutig definiert, so Univ.-Prof. Dr. Albert Wölfler von der Klinischen Abteilung für Hämatologie an der Medizinischen Universität Graz. Damit werden Krankheiten umschrieben, die durch einen Zytokinsturm bedingt sind und zu Organschädigungen führen. Eine typische Erkrankung dieser Gruppe ist die sekundäre hämophagozytische Lymphohistiozytose (HLH), die durch Infektionen, rheumatische oder hämatologische Erkrankungen ausgelöst werden oder iatrogen bedingt sein kann. Erst vor wenigen Jahren erstmals beschrieben wurde das sogenannte VEXAS-Syndrom, das ebenfalls in diese Gruppe gehört, dabei aber einen gänzlich anderen Pathomechanismus aufweist.
Hämophagozytische Lymphohistiozytose (HLH)
Bei der HLH muss zwischen primären undsekundären Formen unterschieden werden. Die primäre HLH tritt zumeist bei Kindern auf und hat in vielen Fällen einen monogenetischen Hintergrund. Die auch als HLH-Spektrum-Erkrankungen bezeichneten sekundären HLH können unterschiedliche Ursachen wie Infektionen, Malignome, Autoimmunität bzw. Autoinflammation haben. Es besteht eine Überschneidung mit der Sepsis, bestimmte immunaktivierende Therapien (CAR-T-Zellen, T-Zell-Engager und Checkpoint-Inhibitoren) werden ebenfalls mit HLH in Verbindung gebracht. Pathophysiologisch ist die HLH durch Überaktivität von natürlichen Killerzellen (NK-Zellen) und zytotoxischen T-Zellen charakterisiert, die jedoch gleichzeitig eine suboptimale Clearance ihrer Targetzellen zeigen. Dadurch kommt es zur persistierendenImmunaktivierung und zur verstärkten Produktion proinflammatorischer Zytokine. Der Zytokinsturm führt zur Aktivierung von Monozyten und Makrophagen. Diese wiederum wandern in die verschiedenen Gewebe und bewirken Spleno- und Hepatomegalie, Lymphadenopathie sowie im Knochenmark die namensgebende histiozytische Phagozytose von Blutzellen. Zu diesen Symptomen kommen typischerweise noch Fieber und eine Reihe von Auffälligkeiten im Labor, wie Zytopenie, erhöhte Triglyzeride, massiv erhöhtes lösliches CD25 (IL-2-Rezeptor) sowie erhöhtes Serum-Ferritin. Bei einer Biopsie des Knochenmarks werden Hämophagozytose, Histiozyten-Hyperplasie und Hyperzellularität erkennbar.1
Vielfältige, auch iatrogene Genese
Ätiologisch kann hinter einer HLH eine Reihe von Krankheitserregern stehen, wobei dem Epstein-Barr-Virus besondere Bedeutung zukommt. Aber auch Herpes-simplex-Virus, Zytomegalievirus, SARS-CoV-2, verschiedene Bakterien sowie Pilze und Parasiten können eine HLH verursachen. Unter den Malignomen kommen vor allem Lymphome und Leukämien infrage. Häufig sind T-Zell-Lymphome mit HLH vergesellschaftet. Unter den Autoimmunerkrankungen sind vor allem der systemische Lupus erythematodes, die rheumatoide Arthritis, chronisch-entzündliche Darmerkrankungen sowie der Morbus Still des Erwachsenen mögliche Auslöser einer HLH. Auch Medikamente können selten eine HLH auslösen. Wölfler nannte einige Antiepileptika und Antibiotika sowie die Immun-checkpoint-Inhibitoren und wies auf einen besonderen Zusammenhang mit der CAR-T-Zell-Therapie hin, die auf gentechnisch veränderten T-Zellen mit synthetischen antigenspezifischen Rezeptoren basiert und in der Hämatoonkologie zunehmend an Bedeutung gewinnt.2
Nach Infusion der CAR-T-Zellen kommt es häufig zu einer überschießenden Immunreaktion mit Zytokin-Freisetzungssyndrom („cytokine release syndrome; CRS). Dieses ist meist transient und gut beherrschbar. Allerdings kann mit Verzögerung eine abermalige Verschlechterung mit Zeichen systemischer Inflammation auftreten. HLH-like-Syndrome treten um den Tag 10 nach der Infusion sowie als Late-onset-Form nach dem Tag 20 auf. Die Häufigkeit hängt von der zu behandelnden Erkrankung sowie von den eingesetzten Zellen ab, liegt mit den mittlerweile etablierten Therapien aber bei bis zu 10%. Das ist bedeutsam, weil ein HLH-like-Syndrom in dieser Situation bei starker Ausprägung mit einer hohen Mortalität verbunden ist, so Wölfler: „Intensivpflichtige Patien-t:innen zeigen eine hohe Sterblichkeit, wobei die genauen pathogenetischen Zusammenhänge noch nicht bekannt sind.“ Mit CAR gegen CD22 dürfte das Risiko etwas höher sein als mit den aktuell gebräuchlichen, gegen CD19 gerichteten Therapien. Jedenfalls trägt das HLH-like-Syndrom entscheidend zur „Non-relapse“-Mortalität bei CAR-T-Zell-Therapien bei.3
Kortikosteroide nach wie vor bei HLH unverzichtbar
In der Therapie der HLH bildet Dexamethason nach wie vor das „Backbone“. Die Therapie wird mit hoher Dosis begonnen und über acht Wochen, bei gutem Ansprechen auch schneller, ausgeschlichen.
Darüber hinaus bestehen mittlerweile aber auch Möglichkeiten einer gewissen Individualisierung der Therapie. Zur Verfügung stehen Immunglobuline, der gegen den IL-1-Rezeptor gerichtete Antikörper Anakinra sowie seit Kurzem der Januskinase(JAK)-Inhibitor Ruxolitinib.4 Eine an der Medizinuniversität Graz durchgeführte Studie zeigte bei intensivpflichtigen Patient:innen mit HLH unter Therapie mit Dexamethason, Immunglobulinen und Ruxolitinib einen raschen Rückgang der Entzündungsparameter innerhalb von zehn Tagen. Das Gesamtüberleben nach sechs Monaten lag bei 86%.5 Allerdings wies Wölfler auf die insgesamt schlechte Datenlage hin, zumal ausschließlich Fallserien und keine kontrollierten Studien zur optimalen Therapie der HLH vorliegen.
VEXAS: erworbene monogenetische Erkrankung
Ein erst im Jahr 2020 erstbeschriebenes Hyperinflammationssyndrom wird mit demAkronym VEXAS bezeichnet, dieses steht für
-
V: Vakuolen (diese werden häufig in Zellen aus Knochenmarkbiopsien von Patient:innen mit VEXAS-Syndrom gefunden);
-
E: E1-Enzym (Ubiquitin aktivierendes Enzym, für das das bei VEXAS-Betroffenen mutierte Gen UBA1 codiert);
-
X: X-chromosomal (das UBA1-Gen liegt auf dem X-Chromosom, Männer sind häufiger betroffen);
-
A: autoinflammatorisch;
-
S: somatisch (Es handelt sich um somatische Mutationen, die im Laufe des Lebens erworben werden. VEXAS ist keine hereditäre Erkrankung.)
Die UBA1-Mutation bewirkt, dass die Isoform des Enzyms UBA1-B nicht mehr gebildet werden kann. Dies hat zur Folge, dass der Abbau von zytoplasmatischen Proteinen über das Proteosom gestört ist und diese in den Zellen akkumulieren. Dies führt zur Ausschüttung proinflammatorischer Zytokine und letztlich zu chronischer Inflammation mit Multisystembeteiligung.6,7 Zusätzlich kommt es zu gestörter Blutbildung mit Bevorzugung der myeloischen Reihe und Entwicklung einer Anämie und Thrombozytopenie.8
Langfristige Remissionen unter Therapie sind möglich
Die resultierenden, zumeist inflammatorischen klinischen Manifestationen können sämtliche Organsysteme und Körperregionen betreffen. Hautveränderungen sind ebenso häufig wie Fieber und Infiltrate in der Lunge. Typisch ist eine Chondritis des Ohrknorpels, so Wölfler. Auch das Thromboserisiko ist erhöht.9 Auch in der Therapie von VEXAS sind Glukokortikoide unverzichtbar. Darüber hinaus bewähren sich Ruxolitinib und der gegen den IL-6R-gerichtete Antikörper Tocilizumab sowie der IL-1-beta-Inhibitor Canakinumab.10 Bei Anakinra treten hingegen Probleme mit der Verträglichkeit auf, so Wölfler. Eine weitere therapeutische Option ist Azacitidin, ein synthetisches Analogon des Nukleosids Cytidin, das aufgrund seiner hypomethylierenden Wirkung bei verschiedenen malignen Erkrankungen des blutbildenden Systems eingesetzt wird. Mit Azacitidin lassen sich langfristige Remissionen erreichen, wobei das Medikament in vierwöchentlichen Zyklen verabreicht werden muss, da sonst ein hohes Rezidivrisiko besteht (Abb.1). Unter Therapie mit Azaci-tidin kommt es zu Verbesserungen des Blutbildes, zu einem Rückgang des Entzündungsmarkers CRP sowie zu einer Abnahme der Allelfrequenz der UBA1-Mutation.11,12
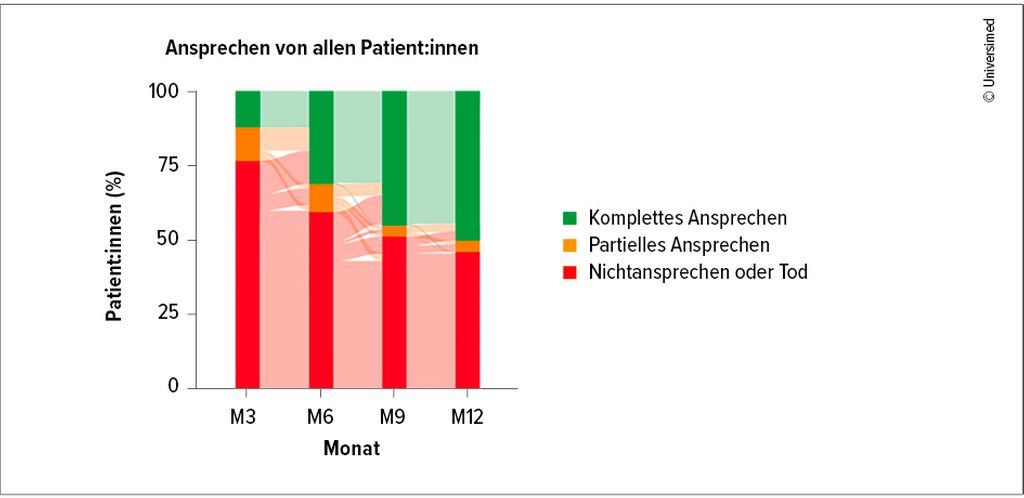
Abb.1: VEXAS-Syndrom: Ansprechen auf Azacitidin (modifiziert nach Jachiet V et al. 2025)12
Quelle:
„Hyperinflammationssyndrome: VEXAS, HLH und andere“, Vortrag von Univ.-Prof. Dr. Albert Wölfler, Graz, im Rahmen des Symposiums „Es ist nicht immer eine Infektionskrankheit! Wichtige DD“ beim 18. ÖIK am 19. März 2026 in Saalfelden
Literatur:
1 Hsu JI et al.: Hemophagocytic lymphohistiocytosis in adults. Blood 2026; 147(10): 1037-47 2 Johnson WT et al.: How I treat HLH-like toxicities following immune effector cell therapy. Blood2026; doi: 10.1182/blood.2025032352 3 Rejeski K et al.: Noncanonical and mortality-defining toxicities of CAR T cell therapy. Nat Med 2025; 31(7): 2132-46 4 La Rosée P, Machowicz R: Hemophagocytic lymphohistiocytosis: do we have a solution for TMI (too much inflammation)? Hematology Am Soc Hematol Educ Program 2025; 2025(1): 206-14 5 Scholz L et al.: Ruxolitinib, iv immunoglobulin, and high-dose glucocorticoids for critically ill adults with secondary hemophagocytic lymphohistiocytosis: a single-center observational pilot study. Crit Care Explor 2024; 6(2): e1046 6 Sakuma M et al.: UBA1 dysfunction in VEXAS and cancer. Oncotarget 2024; 15: 644-58 7 Corrao S et al.: VEXAS Syndrome: Genetics, gender differences, clinical insights, diagnostic pitfalls, and emerging therapies. Int J Mol Sci 2025; 26(16): 7931 8 Molteni R et al.: Mechanisms of hematopoietic clonal dominance in VEXAS syndrome. Nat Med 2025; 31(6): 1911-24 9 Grayson PC et al.: VEXAS syndrome. Blood 2021; 137(26): 3591-94 10 Hadjadj J et al.: Efficacy and safety of targeted therapies in VEXAS syndrome: retrospective study from the FRENVEX. Ann Rheum Dis 2024; 83(10): 1358-67 11 Aalbers AM et al.: Long-term genetic and clinical remissions after cessation of azacitidine treatment in patients with VEXAS syndrome. Hemasphere 2024; 8(8): e129 12 Jachiet V et al.: Efficacy and safety of azacitidine for VEXAS syndrome: a large-scale retrospective study from FRENVEX. Blood 2025; 146(12): 1450-61
Das könnte Sie auch interessieren:

Dermatologische Differenzialdiagnosen bei sexuell übertragbaren Infektionen
Hautveränderungen im Genitalbereich können sowohl für Patient:innen als auch für Behandler:innen herausfordernd sein. Anamnese sowie klinische Untersuchung sind oft mit Scham und STI mit ...

20. Münchner AIDS- und Infektiologie-Tage
Seit vielen Jahren sind die Münchner AIDS-Tage ein Ankerpunkt im HIV-Kongresskalender. 2026 lud die Konferenz dabei bereits zum dritten Mal zu einem Gastspiel nach Berlin. Traditionell ...
Gemeinsam oder doch getrennt? Kombinierte Impfung gegen Covid-19 und Pneumokokken
Mit der zunehmenden Anzahl empfohlener Impfungen wächst auch die Herausforderung der Impfbereitschaft. In diesem Zusammenhang bleibt die Frage nach der Wirksamkeit und Sicherheit der ...