
Blickdiagnosen in der Endokrinologie
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
In der Endokrinologie basiert die Diagnosestellung meist auf Laborwerten. Einige Erkrankungen zeigen sich dennoch als Blickdiagnosen oder können mithilfe einfacher klinischer Tests diagnostiziert werden. Im Folgenden werden besonders typische klinische Zeichen bestimmter endokrinologischer Erkrankungen sowie der Weg zur Diagnose beschrieben.
Cushing-Syndrom
Das Cushing-Syndrom, auch als Hyperkortisolismus bezeichnet, ist durch eine zentrale, viszerale Fettverteilung gekennzeichnet, während die Extremitäten schlank bleiben und eine typische proximale Muskelschwäche auftritt. Typische Merkmale sind zudem Striae rubrae (Abb. 1), pergamentartige Haut, Hämatomneigung, ein Mondgesicht, ausgeprägte Nackenpolster (Stiernacken) und eine verzögerte Wundheilung. Betroffene leiden häufig unter Adipositas, arterieller Hypertonie und (Prä-)Diabetes, depressiver Verstimmung, peripheren Ödemen, Akne, Hirsutismus und Zyklusunregelmässigkeiten. Mögliche Komplikationen umfassen Thrombosen, Infekte, Osteoporose, Entwicklung eines metabolischen Syndroms und eine insgesamt deutlich erhöhte kardiovaskuläre Mortalität.
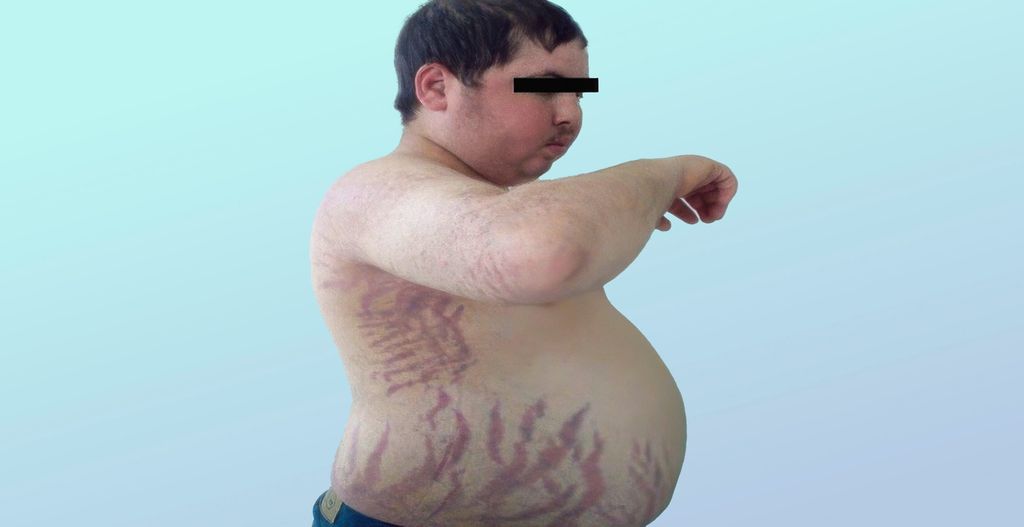
Abb. 1: Typische kutane Merkmale beim Cushing-Syndrom: Striae rubrae, pergamentartige Haut (aus: El Aziz S et al.: Apport de l’orientation clinique dans le diagnostic des hypertensions arterielles endocriniennes. Pan African Medical Journal 2014; 18: 171. Lizensiert unter Creative Commons Attribution 4.0 International [CC BY 4.0])
Die häufigste Ursache ist ein exogener, d.h. ein Adrenocorticotropin(ACTH)-unabhängiger Hyperkortisolismus, der typischerweise nach Langzeittherapie mit Glukokortikoiden entsteht. Das wesentlich seltenere endogene Cushing-Syndrom resultiert aus einer endogen gesteigerten Cortisol- oder ACTH-Produktion, beispielsweise bei Hypophysen- oder Nebennierenadenomen oder auch im Rahmen eines paraneoplastischen Syndroms.
Der diagnostische Goldstandard ist der Dexamethason-Hemmtest. Hierbei nimmt der Patient um 23 Uhr 1mg Dexamethason ein. Liegt der Cortisolspiegel am nächsten Morgen um 8 Uhr ≥50nmol/l, spricht dies für eine erhöhte Cortisolausschüttung. Weitere Analysen, wie die Messung der freien Cortisolausscheidung im 24-Stunden-Urin oder das Mitternachtscortisol im Speichel an drei verschiedenen Tagen, bestätigen die Diagnose.
Klinische Zeichen auf einen Blick
Cushing-Syndrom: | Muskelschwäche, Striae rubrae, Hämatome, Mondgesicht |
Akromegalie: | Vergröberung der Gesichtszüge/Akren, Kopfschmerzen, Schwitzen, metabolisches Syndrom |
Morbus Basedow: | endokrine Orbitopathie, Palpitationen, Schwitzen, Schlafstörungen |
Hypothyreose: | periorbitale Gesichtsschwellung, Bradykardie, Kälteintoleranz |
Morbus Addison: | Hyperpigmentierung der Haut, Schwäche, Gewichtsverlust, Hypotonie |
Die Therapie richtet sich nach der zugrunde liegenden Ursache: Hypophysen- oder Nebennierenadenome werden primär operativ entfernt. Zudem sind Thrombose- und Infektprophylaxe essenziell. Medikamentöse Therapieoptionen stellen Kortikosteroid-Synthese-Hemmer wie Ketoconazol, Metyrapon oder Osilodrostat dar.
Akromegalie
Die typischen klinischen Zeichen einer Akromegalie fallen oft erst verspätet auf, wenn es bereits zu einer deutlichen Vergröberung der Gesichtszüge mit Wachstum des Unterkiefers (Abb. 2), Abweichen der Zähne, Makroglossie und typischerweise Vergrösserung von Händen (Abb. 3) und Füssen gekommen ist. Allerdings leiden Betroffene meist schon lange vorher unter Symptomen wie Kopf- und Gelenkschmerzen, vermehrtem Schwitzen, einem metabolischen Syndrom und gegebenenfalls einem Hypogonadismus. Ein beidseitiges Karpaltunnelsyndrom in jungen Jahren kann ebenfalls den klinischen Verdacht einer Akromegalie wecken.
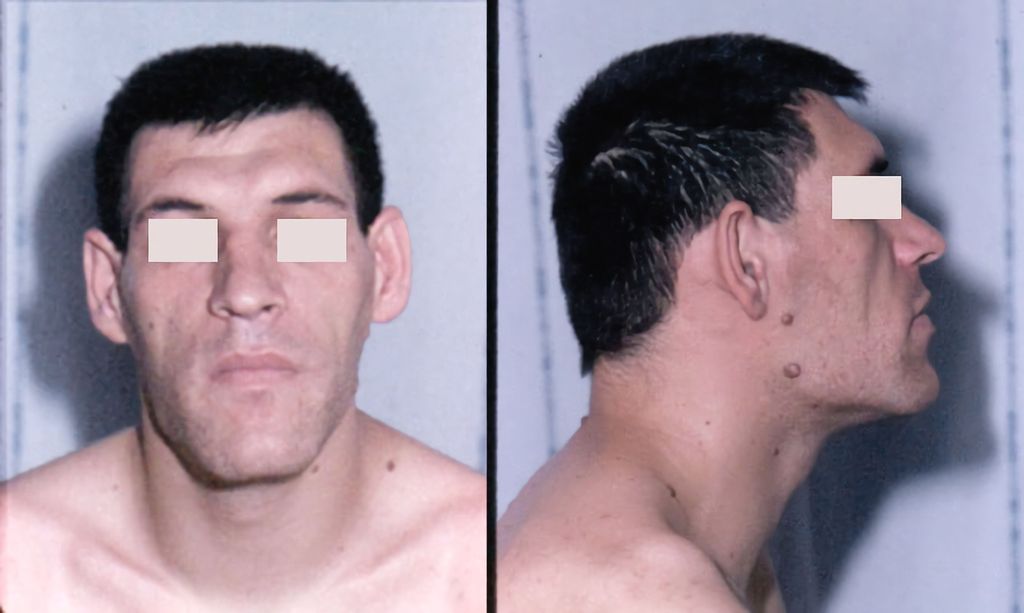
Abb. 2: Typische Gesichtszüge bei Akromegalie (aus: Chanson P, Salenave S: Acromegaly. Orphanet J Rare Dis 2008: 3; 17. Lizensiert unter Creative Commons Attribution License 2.0 Generic [ https://creativecommons.org/licenses/by/2.0 ])

Abb. 3: Stark vergrösserte Hand bei Akromegalie
Die Ursache der Akromegalie sind meist Hypophysenadenome, die zu einer Überproduktion von Wachstumshormon (GH) führen. Dies führt in der Leber zur vermehrten Bildung von «insulin-like growth factor 1» (IGF-1), wodurch Knochen, Muskeln und Organe vermehrt wachsen. Zudem kann es zu einer gestörten Glukosetoleranz, einem metabolischen Syndrom und psychischen Beschwerden kommen. Betroffene haben ausserdem ein erhöhtes Risiko für Kolonpolypen und Darmkrebs.
Die Diagnose wird durch ein unzureichend supprimiertes Wachstumshormon im oralen Glukosetoleranztest (75g Glukose) gestellt. Die Therapie erfolgt in erster Linie durch die chirurgische Entfernung des Hypophysenadenoms. Alternativ oder ergänzend können Medikamente wie Somatostatin-Analoga (Octreotid, Pasireotid), Wachstumshormon-Rezeptorblocker (Pegvisomant) oder Dopamin-Agonisten (Cabergolin) eingesetzt werden.
Morbus Basedow
Auf den ersten Blick fallen Patienten mit M. Basedow durch eine Struma (Abb. 4), einen Exophthalmus (Abb. 5) und eine Lidretraktion auf. Die Augen sind typischerweise gerötet und tränen, und die Patienten berichten von Doppelbildern. Weitere typische Symptome des M. Basedow sind Palpitationen, Gewichtsverlust, vermehrtes Schwitzen und sehr typisch Schlafstörungen. Die Erkrankung wird durch Thyreotropin-Rezeptor-Autoantikörper (TRAK) ausgelöst, die die Produktion von Schilddrüsenhormonen stimulieren. Die Diagnose basiert auf einem erniedrigten Thyreotropin (TSH), bei gleichzeitig erhöhtem freiem Thyroxin (fT4), Trijodthyronin (fT3) und positiven TRAK. Letztere zeigen eine hohe Sensitivität und Spezifität für den M. Basedow. Zusätzlich können im Rahmen der Hyperthyreose erhöhte Transaminasen, eine erhöhte alkalische Phosphatase sowie eine Leukopenie und Anämie auftreten.

Abb. 4: Struma bei Morbus Basedow (aus: www.commons.wikimedia.com. Lizensiert unter Creative Commons Attribution-Share Alike 3.0 Unported, 2.5 Generic, 2.0 Generic and 1.0 Generic)

Abb. 5: Endokrine Orbitopathie bei Morbus Basedow
Die Behandlung des M. Basedow besteht aus Thyreostatika (z.B. Carbimazol) über 12–18 Monate. Das Rezidivrisiko nach Absetzen der Therapie liegt bei circa 50%. Eine seltene, aber schwerwiegende Nebenwirkung von Thyreostatika ist die Agranulozytose, weshalb zu Therapiebeginn und insbesondere unter hohen Dosen der Thyreostatika eine Kontrolle der Leukozyten und der Neutrophilen erfolgt. Bei einer endokrinen Orbitopathie werden Tränenersatzmittel und ein Rauchstopp empfohlen. In schweren Fällen können eine hoch dosierte Steroidtherapie sowie eine chirurgische Dekompression angezeigt sein. Im Falle von Rezidiven des Morbus Basedow kann eine Radiojodtherapie erwogen werden, wobei jedoch eine bestehende Orbitopathie verschlechtert werden kann. Alternativ kann eine Thyreoidektomie erwogen werden.
Hypothyreose
Patienten mit einer ausgeprägten Hypothyreose können insbesondere im Gesicht periorbital eine teigige Hautschwellung aufweisen (Abb. 6). In schweren Fällen haben sie eine Bradykardie und wirken apathisch. Weitere Zeichen sind Kälteintoleranz, Gewichtszunahme, Haarverlust und Zyklusstörungen sowie abgeschwächte Reflexe. Die Diagnose erfolgt durch die Bestimmung der TSH- und fT4-Spiegel. Bei autoimmuner Genese können erhöhte Thyreoperoxidase-Antikörper (TPO) nachweisbar sein. Begleitend können in schweren Fällen eine Hyponatriämie, Anämie und Hypercholesterinämie auftreten. Eine seltene, aber schwere Komplikation ist das Myxödem-Koma mit einer Mortalität von bis zu 15%.
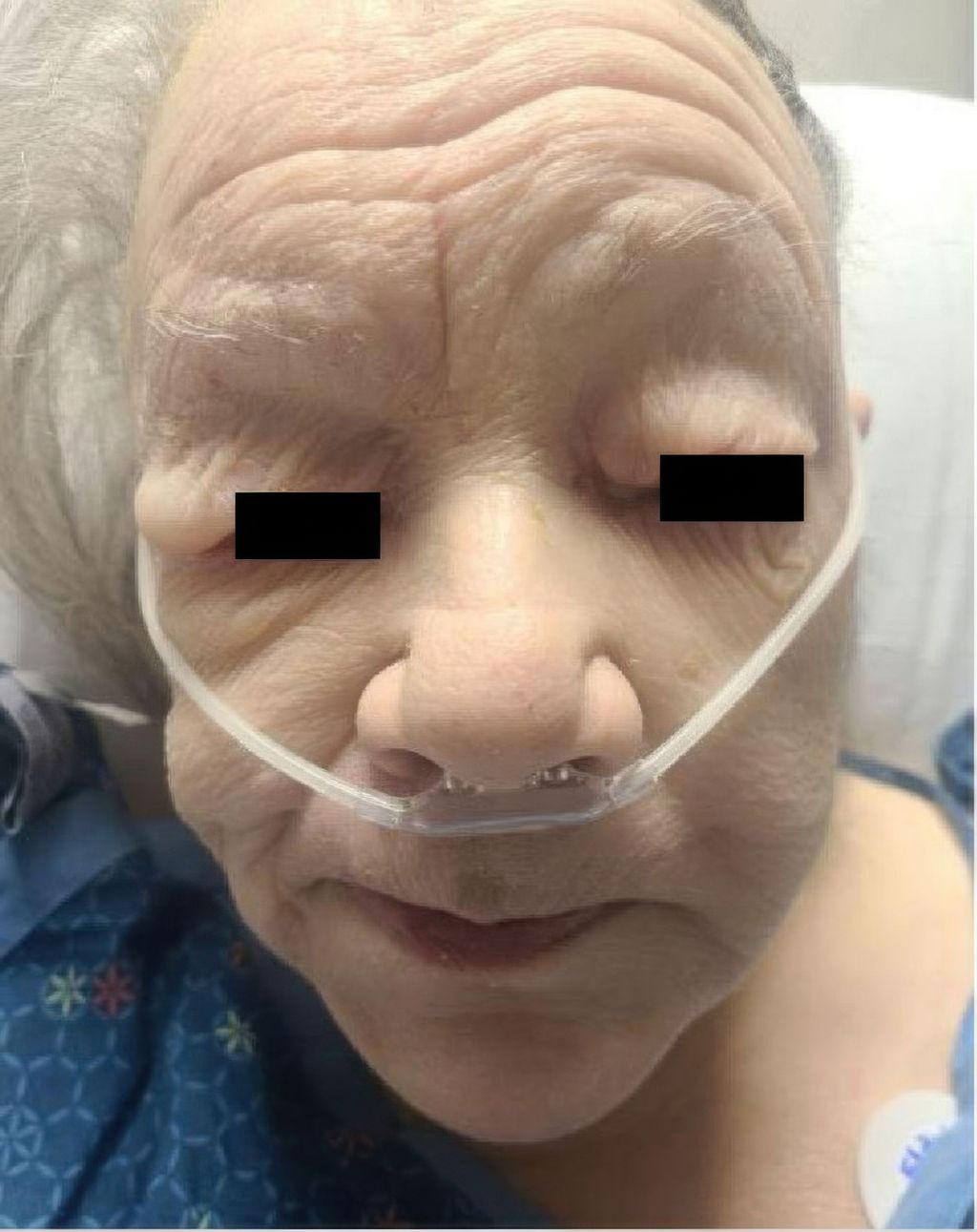
Abb. 6: Myxödem bei Hypothyreose (aus: Ponnapalli A et al.: Pericardial effusion uncovering underlying hypothyroidism. Clin Case Rep 2021; 9: 1816-8. Lizensiert unter Creative Commons Attribution License)
Die Therapie erfolgt mit Levothyroxin in einer Zieldosis von 1,6µg/kg/d, wobei bei älteren und kardial vorerkrankten Patienten die Dosis schrittweise aufdosiert werden sollte. Bei einem Myxödem oder bei Verdacht auf weitere Hormonausfälle muss erst eine Nebenniereninsuffizienz ausgeschlossen werden, bevor mit Levothyroxin (300–500µg i.v.) begonnen wird, um eine Addison-Krise zu verhindern. Bei subklinischer Hypothyreose wird ab einem TSH-Wert >10mU/l ein Therapiebeginn empfohlen.
Bei akuter Krankheit, z.B. im stationären Setting, können die Schilddrüsenwerte nicht sicher interpretiert werden («non-thyroidal illness syndrome»).
Morbus Addison
Die primäre Nebennierenrindeninsuffizienz zeigt sich unter anderem durch eine Hyperpigmentierung der Handinnenfalten (Abb. 7), Narben und Mundschleimhaut. Diese entsteht durch das Prohormon des ACTH (POMC). Die Patienten klagen über ausgeprägte Schwäche, Adynamie, Gewichtsverlust, Hypotonie. Auch ein verminderter Insulinbedarf bei Typ-1-Diabetes oder neue Hypoglykämien können klinische Zeichen sein. Typische Laborveränderungen sind Elektrolytentgleisungen wie Hyponatriämie und Hyperkaliämie.

Abb. 7: Hyperpigmentierung der Handinnenfalten bei Morbus Addison (aus: Barts Health NHS Trust Archives SBHB/MU/14/27/9/2. Photo number: L0061738. Lizensiert unter Creative Commons Attribution 4.0 International license)
Eine mögliche Ursache für den M. Addison ist die Autoimmunadrenalitis, in selteneren Fällen auch ein Nebennierenrindenkarzinom, Metastasen, Blutungen, infektiöse Erkrankungen (z.B. Tuberkulose, Zytomegalievirusinfektion) sowie infiltrative Erkrankungen wie Lymphome oder die Amyloidose. Ein morgendlicher Cortisol-Wert <150nmol/l bestätigt die Diagnose einer Nebenniereninsuffizienz, während ein Wert >300nmol/l die Diagnose ausschliesst. Bei Verdacht auf M.Addison sollte zeitnah mit einer Hydrocortison-Substitution begonnen werden, um eine potenziell lebensbedrohliche Addison-Krise zu verhindern. Die Erhaltungstherapie besteht aus der Hydrocortison-Substitution (bspw. 15–5–0mg), ergänzt durch eine Stressprophylaxe und umfassende Schulung der Patienten. Bei begleitendem Aldosteronmangel wird die Therapie durch Fludrocortison ergänzt
Literatur:
● Fleseriu M et al.: Lancet Diabetes Endocrinol 2021; 9: 847-75 ● Nieman LK et al.: J Clin Endocrinol Metab 2015; 100: 2807-31 ● Fassnacht et al.: Eur J Endocrinol 2016; 175: 1-34 ● Fleseriu M et al.: Pituitary 2020; 24: 1-13 ● Giustina A et al.: J Clin Endocrinol Metab 2019; 105: dgz096 ● Bartalena I et al.: Eur J Endocrinol 2021; 185: 43-67 ● Jonklaas J et al.: Thyroid 2021; 31: 156-82 ● Beuschlein F et al.: J Clin Endocrinol Metab 2024; 109: 1657-83 ● Vita JA et al.: Am J Med 1985; 78: 461-6 ● Gravholt CH et al.: Eur J Endocrinol 2024; 190: 53-151 ● Zitzmann M et al.: Andrology 2020; 9: 145-67
Das könnte Sie auch interessieren:

Diabetes erhöht das Sturzrisiko deutlich
Eine dänische Studie kommt zu dem Ergebnis, dass sowohl Patienten mit Typ-1- als auch Patienten mit Typ-2-Diabetes öfter stürzen und häufiger Frakturen erleiden als Menschen aus einer ...
Notfall Diabetische Ketoazidose: Leitliniengerechtes Handeln kann Leben retten
Akute Stoffwechselentgleisungen können lebensbedrohlich sein und erfordern eine rasche und leitliniengerechte Diagnostik und Therapie. Pathogenese, Klinik, typische Befunde und die ...

Wie oft wird Diabetes nicht oder spät erkannt?
Im Allgemeinen wird von einer hohen Dunkelziffer an Personen mit undiagnostiziertem Typ-2-Diabetes ausgegangen. Ein Teil davon sind von Ärzten „übersehene“ Fälle. Eine von der University ...