Behçet-Syndrom: von der oft schwierigen Diagnose zur Therapie
Das Behçet-Syndrom ist aufgrund genetischer Prädisposition vor allem in den Regionen entlang der Seidenstrasse verbreitet. Die chronische entzündliche Multisystemerkrankung weist eine heterogene Krankheitsausprägung auf, weshalb die Diagnose komplex ist: Sie beruht auf dem Ausschluss zahlreicher alternativer Diagnosen. In der klinischen Praxis gilt es daher einigeszu beachten.
Keypoints
-
Das Symptom Ulzera ist vielfältig, das Behçet-Syndrom jedoch selten.
-
Für die Diagnose können Klassifikationskriterien der ISG und die International Criteria for Behçet’s Disease (ICBD) hilfreich sein.
-
Mögliche Therapien orientieren sich an den jeweiligen Organmanifestationen.
-
Vor allem bei kutanen Manifestationen sollten Dermatolog:innen an das Behçet-Syndrom denken.
Das Behçet-Syndrom ist in wesentlichen Industrienationen eine seltene Erkrankung: Die Prävalenz beträgt 4,03/100000 Einwohner:innenin der Gesamtbevölkerung der Schweiz. Betrachtet man jedoch ausschliesslich Bevölkerungsgruppen, deren Herkunftsland mit hohen Prävalenzzahlen assoziiert ist, wie bspw.die Türkei oder der Iran, so steigt diese Zahl auf 19,5/100000.1,2 «Bis dato ist das Behçet-Syndrom pathophysiologisch noch nicht gut verstanden», erläuterte PD Dr. med. Christof Iking-Konert, Chefarzt der Abteilung Rheumatologie am Stadtspital Zürich. Es ist an genetische und umweltspezifische Faktoren geknüpft, die zu den regionalen Schwankungen der Prävalenz und der Krankheitsausprägung beitragen.1,2 Neben Schadstoffbelastung sind möglicherweise Infektionserreger wie das Herpes-simplex-Virus sowie Streptococcus sanguinis mit dem Ausbruch der Erkrankung assoziiert.3 «Mit HLA-B51 haben wir einen genetischen Marker, der zwar prädisponierend für das Behçet-Syndrom, aber aufgrund seiner Häufigkeit in den Hochprävalenzregionen nicht für die Diagnose geeignet ist. Weder der Nachweis von HLA-B51 noch das Fehlen beweisenein Behçet-Syndrombzw. schliessen es aus und leider existiert auch kein anderer zuverlässiger serologischer Diagnosemarker», so Iking-Konert.
Herausfordernde Diagnostik
Klassifiziert wird das Behçet-Syndrom als variable Gefässerkrankung, da es sowohl grosse als auch kleine Gefässe betreffen kann. Zudem finden sich viele entzündliche Manifestationen auf der venösen Seite – eine Besonderheit dieses Syndroms. Die Dermatologie ist eine prädestinierte Disziplin, um dieErkrankung zu diagnostizieren: In nahezu 100% der Fälle weisen die Betroffenen rezidivierende orale Ulzera in Form von Aphthen auf, in 80% genitale Ulzera.4,5 Damit gelten diese als die Kardinalsymptome des Behçet-Syndroms.6 «Die Ursachen oraler Ulzerationen sind jedoch vielfältig und das Behçet-Syndrom ist selten», so der Experte. Als mögliche Differenzialdiagnosen kommen unter anderem rheumatische, hämatologische, autoinflammatorische, gastrointestinale und dermatologische Erkrankungen und Infektionen infrage.
Rund die Hälfte der Behçet-Patient:innen entwickelt daneben eine Augenbeteiligung. Diese tritt bei den meisten Betroffenen innerhalb von zwei Jahren nach der Erstuntersuchung ein. Vor allem bei Männern kann diese okuläre Manifestation gravierend verlaufen, bis hin zum Verlust der Sehkraft.7 Darüber hinaus sind unter anderem die Gelenke, der Gastrointestinaltrakt, die arteriellen wie venösen Gefässe und das zentrale Nervensystem beteiligt (Abb. 1).
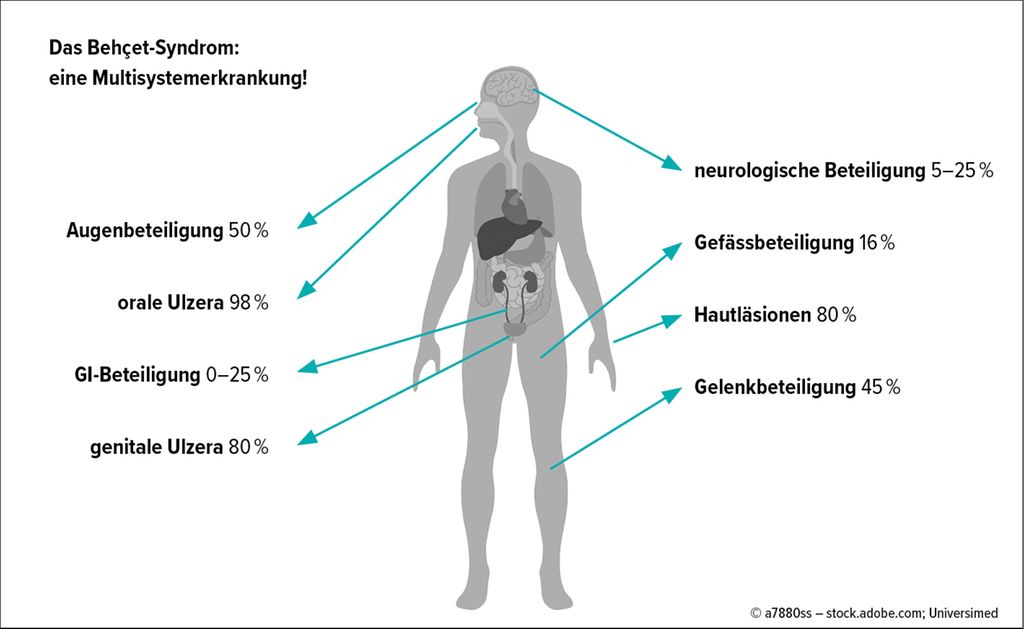
Abb. 1: Manifestationen von Morbus Behçet mit näherungsweisen Prävalenzen (modifziert nach Verity DH et al. 2003 und Barnes CG et al. 2010)4,5
An der Haut zeigen sich neben Aphthen vor allem Akne-ähnliche papulopustuläre Veränderungen, Erythema-nodosum-artige Läsionen sowie Pyodermie. Weiterhin können superfizielle Thrombophlebitiden und Hautulzera auftreten.8,9 Die klinischen Symptome des Behçet-Syndroms haben erhebliche körperliche und psychische Auswirkungen und können zu einer deutlichen Beeinträchtigung der Lebensqualität sowie zu einer erhöhten Morbidität und Mortalität führen.
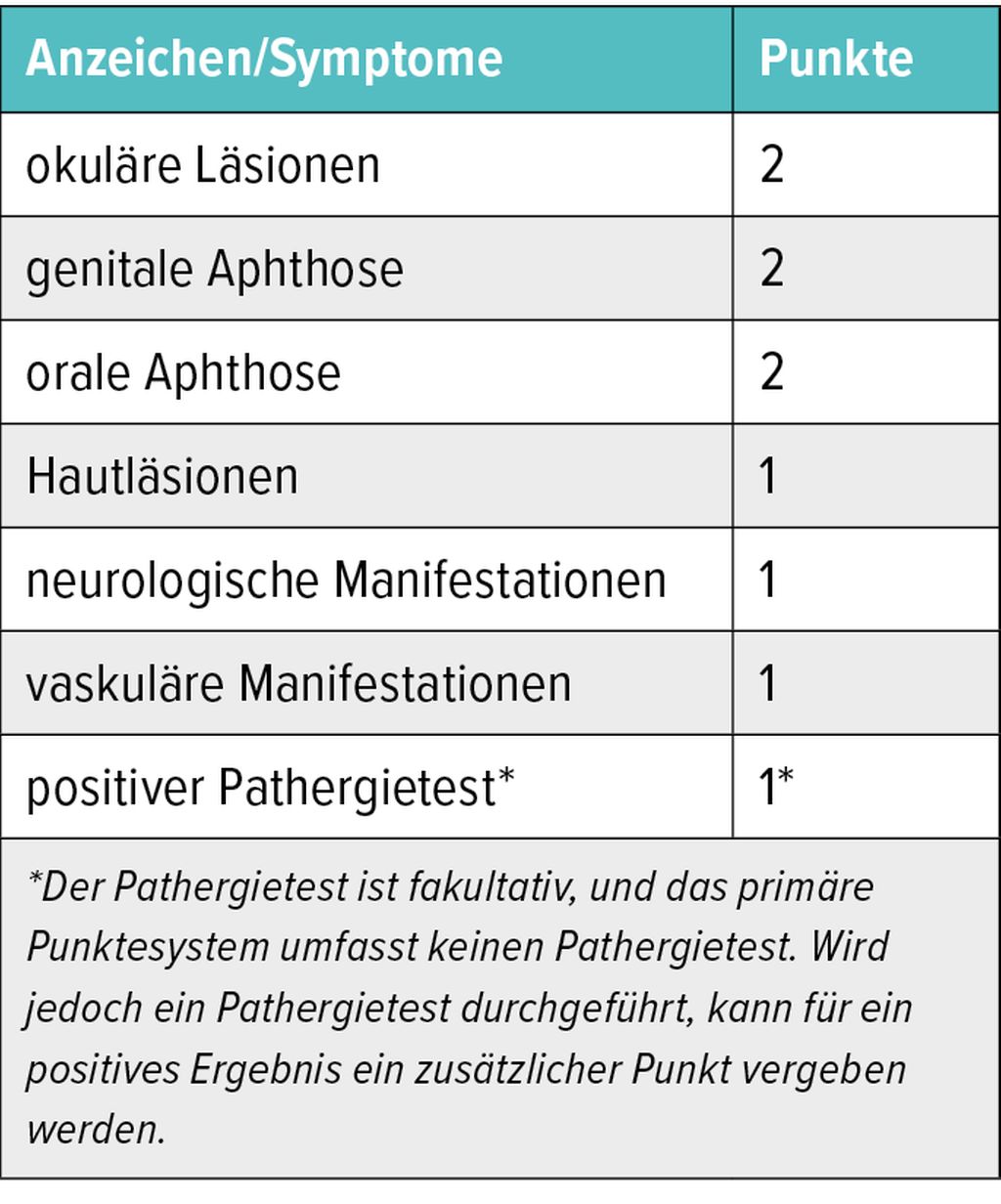
Tab. 1a: Internationale Kriterien des Behçet- Syndroms (ICBD); Punktesystem: ≥ 4 Punkte führen zur Behçet-Diagnose
Die mittlere Dauer vom ersten Auftreten der Symptome bis zur Diagnosestellung beträgt 5,3 Jahre – die Diagnose ist tendenziell langwieriger bei Patient:innenmit ausschliesslich mukokutanen Läsionen (1,13±2,4 Jahre) als bei denen mit schwererwiegender Organbeteiligung (0,88±1,9 Jahre).6 Zur Diagnosefindung können unter anderem die Klassifikationskriterien der Internationalen Studiengruppe (ISG) hilfreich sein.10 Demnach müssen neben rezidivierenden oralen Ulzera mindestens zwei der folgenden Kriterien erfüllt sein: rezidivierende genitale Ulzera, Augenläsionen, Hautläsionen oder ein positives Ergebnis im Pathergietest. Seit 2014 bietendie ICBD (International Criteria for Behçet’s Disease) mit einem Punktesystem Unterstützung bei der Diagnosestellung (Tab. 1a).11 «Mit diesen Kriterien können Symptome zu einem bestimmten Grad der Wahrscheinlichkeit des Vorliegens eines Behçet-Syndroms aufaddiert werden», so Iking-Konert.
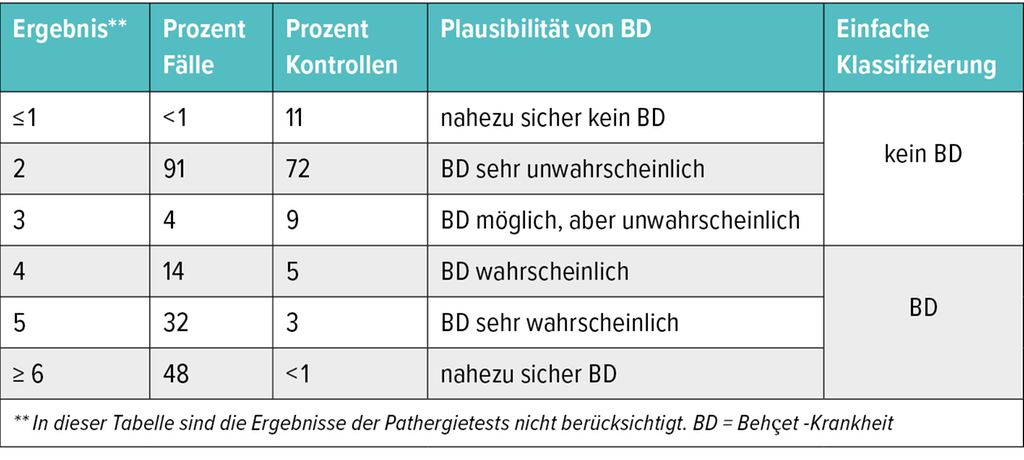
Tab. 1b: Verteilung der Punkte in Fällen und Kontrollen (modifiziert nach Villiger RA et al. 2019)2
Therapie an Organmanifestationen orientieren
Bei der Behandlung des Behçet-Syndroms sollte man sich an den Empfehlungen der European Alliance of Associations for Rheumatology (EULAR) von 2018 orientieren.12 «Ein grosses Problem ist, dass die verfügbare klinische Evidenz zu den verschiedenen Manifestationen des Behçet-Syndroms sehr lückenhaft ist», führte der Rheumatologe aus. Das Wissen basiere auf Registerstudien sowie kleinen, unkontrollierten Fallserien und nur auf sehr wenigen randomisiert-kontrollierten Studien (RCT).
Die Therapieempfehlungen nach EULAR orientieren sich eng an den Organmanifestationen (Tab. 2): Bei kutanen Manifestationen kommen in der Erstlinie Colchicin und lokale Steroide zum Einsatz. Bei okulärer Beteiligung werden Steroide, Azathioprin, Cyclosporin A sowie TNFα-Inhibitoren – Letztere vor allem bei der retinalen Vaskulitis – empfohlen. «Bei den gastrointestinalen Manifestationen behandeln wir an unserem Zentrum analog zu einer chronisch-entzündlichen Darmerkrankung», berichtete Iking-Konert. Liegen neurologische Symptome vor, kommen im Stadtspital Zürich bei älteren Patient:innen häufig Cylophosphamid, bei jüngeren TNFα-Inhibitoren – bevorzugt Infliximab – zum Einsatz. Bei einer Gelenkmanifestation wie bei polyartikulärer Arthritis wird analog zu rheumatoiden Arthritis mit Methotrexat (MTX), Sulfasalazin (SSZ) oder auch TNFα-Inhibitoren therapiert, in Anlehnung an diese Erkrankung und mangels besserer randomisierter Studien für das Behçet-Syndrom.
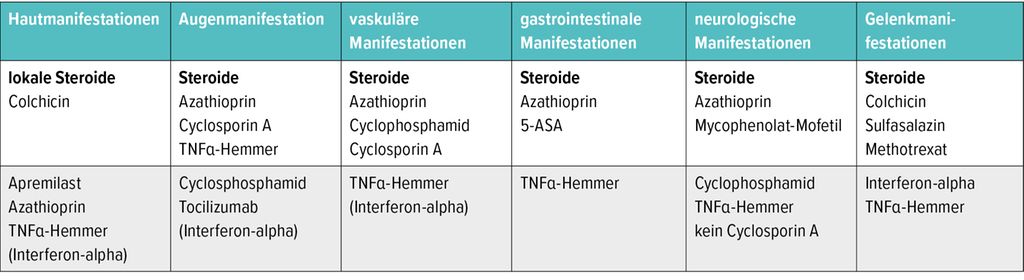
Tab. 2: EULAR-Therapieempfehlungen (Stand: 2018; modifiziert nach Hatemi G et al. 2018)12
Hinsichtlich des Phosphodiesterase-4-Inhibitors Apremilast (Otezla®) wies der Experteauf die recht strengen Limitationen in der Schweiz in dessen Anwendung hin, die nicht einfach zu einzuhalten sind: Das Medikament ist nur für kutane und explizit nicht für neurologische oder andere organbedrohliche Manifestationen oder in Kombinationen mit Biologika erstattungsfähig. In der Zulassungsstudie aus dem Jahr 2019 erhielten 104 Behçet-Patient:innen mit aktiven oralen Ulzera, aber ohne grössere Organbeteiligung 2xtäglich Apremilast 30mg (vs. 103 Patient:innen Placebo).13 Der primäre Endpunkt war die «area under the curve» (AUC) für die Gesamtzahl der oralen Ulzera nach 12 Wochen. In der Apremilast-Gruppe betrug die AUC 129,5 vs. 222,1 für Placebo (p<0,001). «Es halbiert somit die Wahrscheinlichkeit des Auftretens neuer mukokutaner Manifestationen», so Iking-Konert. In der Studie wurde als Nebenwirkungen der Therapie meist nur von leichtem Durchfall, leichter Übelkeit und leichten Kopfschmerzen berichtet.
Abschliessend appellierte der Experte daran, gerade als Dermatolog:innen bei kutanen Manifestationen an diese seltene Erkrankung zu denken, vor allem, wenn die Patient:innen einen entsprechenden familiären Hintergrund aus Hochprävalenzregionen wie dem Mittelmeerraum haben. «Es liegt mutmasslich eher seltener ein Behçet-Syndromvor, wenn man es vermutet hat – zumindest bei Beginn der Erkrankung», so Iking-Konert. Jedoch könne sich das Syndrom im Verlauf noch entwickeln und dementsprechend gelte es die Patient:innen zu informieren und zu schulen.
Quelle:
Digitaler Update-Kurs:«Entzündliche Dermatosen», Vortrag von PD Dr. med. Christof Iking-Konert, Zürcher Dermatologische Fortbildungstage, Juni 2024, Zürich
Literatur:
1 Cho SB et al.: New insights in the clinical understanding of Behçetʼs disease. Yonsei Med J 2012; 53(1):35-42 2 Villiger RA et al: Behçet’s syndrome: clinical presentation and prevalence in Switzerland. Swiss Med Wkly 2019; 149: w20072 3 Alpsoy E: behçet‘s disease: a comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol 2016; 43(6): 620-32 4 Verity DH et al.: Behçet’s disease: from hippocrates to the third millennium. Br J Ophthalmol 2003; 87(9): 1175-83 5 Barnes CG: History and diagnosis. In: Behçet’s syndrome. Springer 2010 6 Alpsoy E et al.: Clinical features and natural course of Behçet’s disease in 661 cases: a multicentre study. Br J Dermatol 2007; 157(5): 901-6 7 Kural-Seyahi E et al.: The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore) 2003; 82(1): 60-76 8 Altenburg A et al.: Epidemiologie und Klinik des Morbus Adamantiades-Behçet in Deutschland. Aktuelle Daten. Ophthalmologe 2012; 109(6): 531-41 9 Bonitsis NG et al.: Gender-specific differences in Adamantiades-Behçet’s disease manifestations: an analysis of the german registry and meta-analysis of data from the literature. Rheumatology (Oxford) 2015; 54(1): 121-33 10 International Study Group for Behçet’s Disease: Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335: 1078-80 11 Davatchi F et al.: The international criteria for behcet’s disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol 2014; 28: 338-47 12 Hatemi G et al.: 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis 2018; 77(6): 808-18 13 Hatemi G et al.: Trial of Apremilast for oral ulcers in Behçet’s syndrome. N Engl J Med 2019; 381(20): 1918-28
Das könnte Sie auch interessieren:

Tägliche Tablette gegen Psoriasis
Die US-Arzneimittelbehörde FDA hat mit Icotrokinra ein orales Medikament gegen Schuppenflechte zugelassen, welches die Rezeptoren für Interleukin-23 (IL-23) hemmt. Eine EU-Zulassung ...
Juveniles Polymyositis/Sklerodermie-Overlap-Syndrom
Die Komplexität von Kollagenosen und Autoimmunerkrankungen stellt oft eine diagnostische und therapeutische Herausforderung dar, da viele Patient:innen nicht nur einer einzigen Entität ...

Funktionalisierung bakterieller Nanocellulose
Chronische Wunden unterschiedlicher Ätiologie, etwa bei arterieller und venöser Insuffizienz oder Diabetes mellitus, prägen die tägliche Wundversorgung.Neben chronischen Wunden verlangen ...