Therapieauswahl nach pulmonalem Inflammations- und Fibrosemuster
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Da Fibrose und Entzündung in unterschiedlichem Ausmaß zu Lungenbeteiligungen bei rheumatologischen Erkrankungen beitragen, werden im klinischen Alltag Methoden gebraucht, um beide Veränderungen zu bestimmen. Neue Studien zeigen, was zielführend ist. Die Therapie muss der individuellen Situation folgen.
Interstitielle Lungenerkrankungen (ILD) präsentieren sich mit sehr heterogenen Veränderungen des Lungenparenchyms. Hierzu gehören Milchglastrübungen, Retikulationen, Traktionsbronchiektasen, Honeycombing, Zysten und Lungendistorsionen, führte Dr. Tim Oqueka, Hamburg, aus. Ein erhöhtes ILD-Risiko tragen beispielsweise Menschen mit rheumatoider Arthritis (RA) oder systemischer Sklerose (SSc).1 Die ILD-Pathophysiologie gliedert sich in 2 Phasen, wobei es in der frühen zu einer chronischen Inflammation kommt, die bei rheumatologischen Erkrankungen oft durch Autoimmunprozesse induziert wird. Auch Umweltfaktoren wie Tabakrauch oder Medikamente können Auslöser sein. In der späten Phase kann sich, unter Bildung von Myofibroblasten, Kollagenablagerung und der Entstehung extrazellulärer Matrix, eine Fibrose entwickeln. Diese kann sich selbst unterhalten, insbesondere bei Autoimmunität oder wenn das auslösende Agens persistiert, wobei die Inflammation bestehen bleiben kann.2
Es gebe eine große Gruppe interstitieller Lungenerkrankungen (ILD), darunter zahlreiche Autoimmunerkrankungen, bei denen auch Entzündungsprozesse wesentlich seien, führte Oqueka weiter aus. So lassen sich zum Beispiel bei einer nicht-spezifischen interstitiellen Pneumonie (NSIP) in histologischen Schnitten viele Entzündungszellen im Interstitium erkennen. Bei der idiopathischen pulmonalen Fibrose (IPF) mit „usual-interstitial-pneumonia“(UIP)-Muster, dem Grundtypus einer progressiven Fibrose, sieht man hingegen eher fleckförmige Fibrosen, Architekturstörungen und Fibroblastenfoci. Bei rheumatologischen Erkrankungen wie der UIP-Kollagenose (UIP-CTD) sind zusätzlich zu den Fibrosezeichen Entzündungszellen vorhanden.4 Dem individuellen Ausmaß der Fibrose und/oder Entzündung müsse man diagnostisch näherkommen, so Oquekas Fazit.
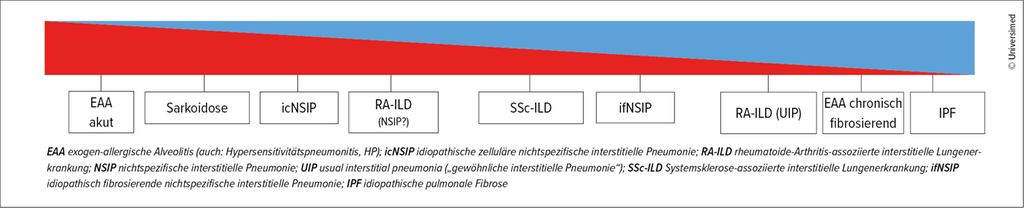
Abb. 1: Das Ausmaß der entzündlichen und fibrotischen Prozesse unterscheidet sich zwischen verschiedenen Erkrankungen. Übergang von entzündlich dominierten zu fibrosedominierten Formen der interstitiellen Lungenerkrankungen von links nach rechts (adaptiert nach Behr J et al. 2023)3
Diagnose der Fibrose
Die Auskultation sei ein sehr einfaches Tool, um eine Fibrose klinisch zu detektieren. Das typische endinspiratorische Knistern habe sich als sehr sensitives und spezifisches Kriterium erwiesen, betonte Oqueka.5 Darüber hinaus kann die Lungenfunktion überprüft werden. Mittelgradige bis schwere Restriktionen weisen auf eine Fibrose hin. Rein entzündungsbedingte Restriktionen können zwar auch auftreten, sind jedoch weniger ausgeprägt. Auch die Diffusion wird als prognostischer Faktor genutzt. Eine Diffusionsstörung könne jedoch bei entzündlichen und fibrotischen Prozessen vorkommen, mahnt Oqueka zur Vorsicht.6
Im Mittelpunkt der bildgebenden Diagnostik steht die High-Resolution-Computertomografie (HRCT). Wichtig ist hier die koronare und sagittale Rekonstruktion der Lunge mit geringer Schichtendicke. Auf Kontrastmittel sollte verzichtet werden, weil sich so das Interstitium besser darstellen lässt. Die Low-Dose-HRCT kommt mit einer um den Faktor 5 bis 10 niedrigeren Dosis aus, genügt zur Beurteilung des Lungengewebes und eignet sich zur Verlaufskontrolle.
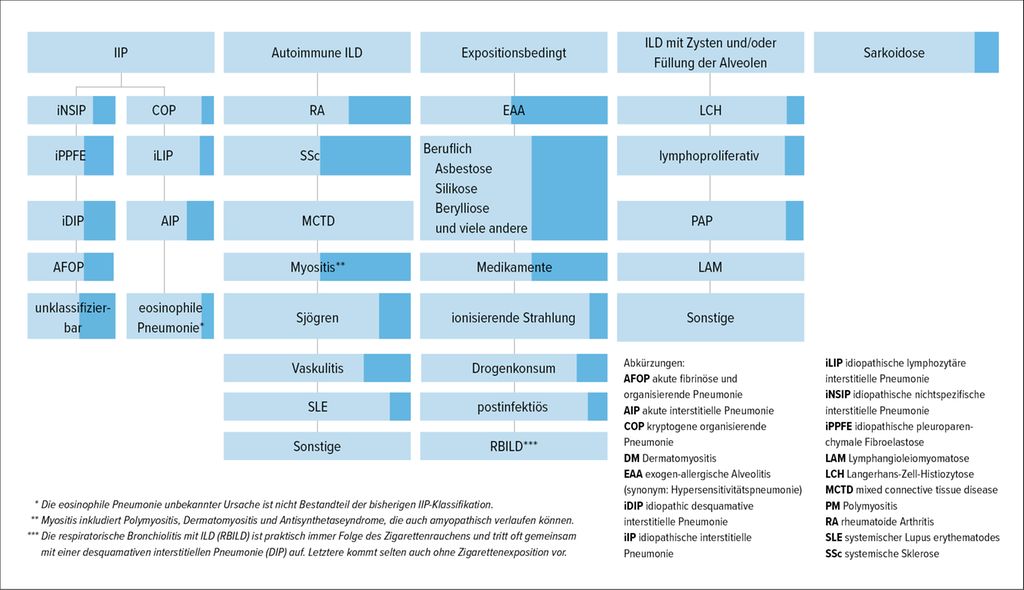
Abb. 2: Übersicht über die interstitiellen Lungenerkrankungen (ILD), die auch als progrediente pulmonale Fibrose (PPF) in Erscheinung treten können. Der dunkelblau gefärbte Anteil des jeweiligen Kästchens repräsentiert den geschätzten Anteil der progredient fibrosierenden Krankheitsverläufe innerhalb der jeweiligen Diagnosegruppe. Die idiopathische pulmonale Fibrose (IPF) bleibt in dieser Darstellung unberücksichtigt (adaptiert nach Raghu G et al.).20
Fibrotische Muster
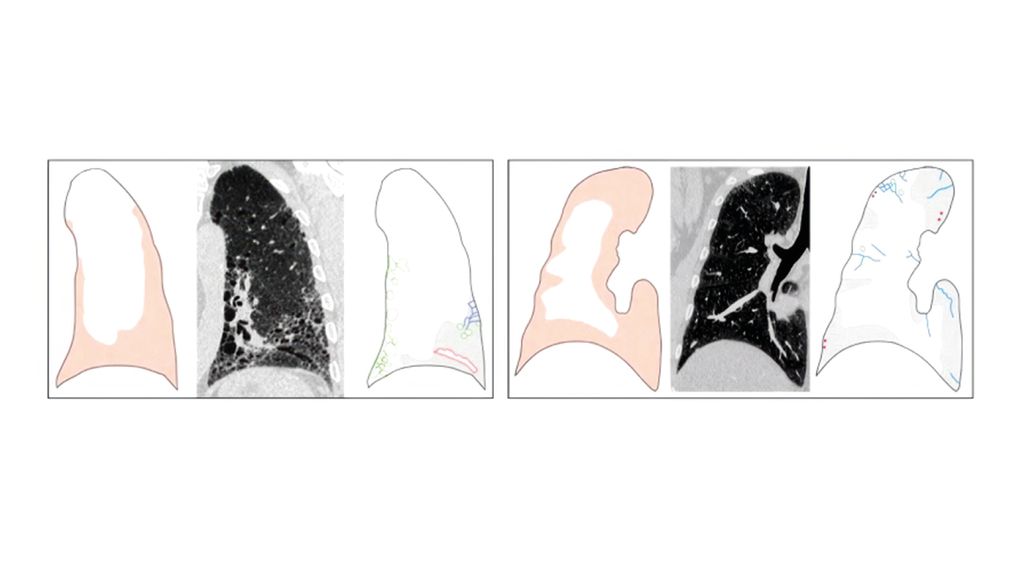
Typische Zeichen für interstitielle Veränderungen sind Architekturstörungen sowie irreguläre Retikulationen (ggf. in Milchglas) im HRCT. Eine Fibrose lasse sich jedoch erst dann eindeutig diagnostizieren, wenn auch Traktionsbronchiektasen und vor allem Honeycombing vorliegen. Es ist wichtig, zwei fibrotische Muster zu differenzieren: die „usual-interstitial“ Pneumonie oder herkömmliche interstitielle Pneumonie von der nichtspezifischen interstitiellen Pneumonie (NSIP), da sich ihre Prognosen unterscheiden, betonte Oqueka. Eine eher basale, periphere Ausprägung mit Traktionsbronchiektasen und Honigwabenmuster kennzeichnet die UIP. Die NSIP hingegen ist diffus über die ganze Lunge verteilt, der periphere Randsaum typischerweise ausgespart (Abb. 3).
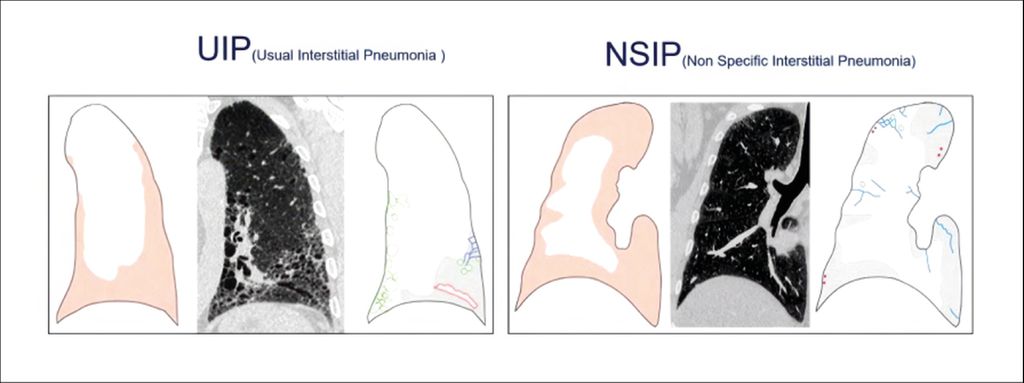
Abb. 3: Typische fibrotische Muster: UIP und NSIP (adaptiert nach Müller-Mang C et al.)7
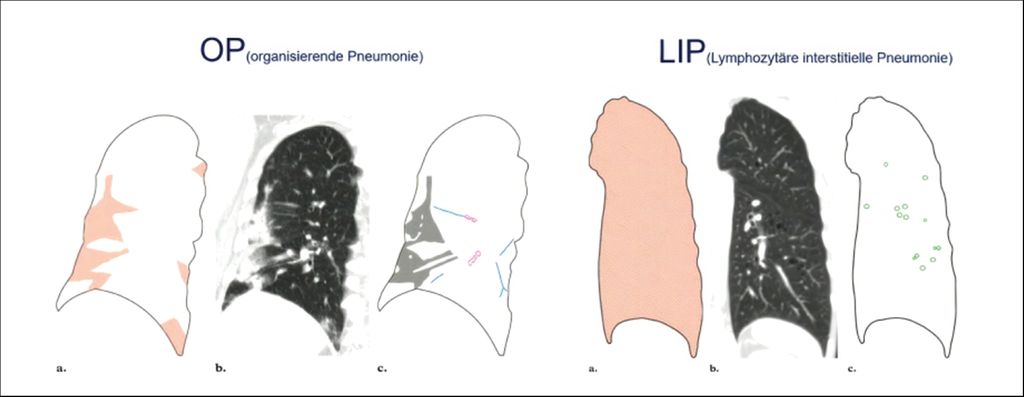
Abb. 4: Typische inflammatorische Muster: OP und LIP (adaptiert nach Müller-Mang C et al.)7
Inflammatorische Muster
Milchglastrübungen ohne irreguläre Retikulationen oder Traktionsbronchiektasen sind typische Entzündungszeichen. Außerdem gehören Konsolidierungen dazu, ebenfalls ohne begleitende Traktionsbronchiektasen, sowie Noduli, und zwar insbesondere Milchglasnoduli.
Bei der organisierenden Pneumonie (OP), einem weiteren für die Rheumatologie wichtigen Muster, sieht man hingegen peripher typische wolkige Infiltrate mit Bronchopneumogramm. Diese rein inflammatorische Erkrankung spricht auf eine antiinflammatorische Therapie sehr gut an. Zudem ist die lymphozytäre interstitielle Pneumonie (LIP) mit Zysten und Milchglastrübungen bei rheumatologischen Erkrankungen nicht selten. Bei Milchglastrübungen handelt es sich um Areale, die röntgendichter sind als andere, jedoch Gefäße und Bronchialwände nicht maskieren. Sie können infolge von Entzündungen auftreten, jedoch auch Ausdruck einer Verdichtung im Aveolarraum sein, eines verdickten Interstitiums oder seiner Destruktion. Mittels einer Verlaufsbildgebung lässt sich dies differenzieren, so Oqueka weiter: Wandern Milchglastrübungen, spricht dies sehr für eine Entzündung, sind sie konstant vorhanden, besteht meist schon eine Fibrose.
Bronchoalveoläre Lavage
In der aktuellen Leitlinie zur Abklärung interstitieller Lungenerkrankungen wird an vielen Stellen die bronchoalveoläre Lavage (BAL) verlangt, sie erhöht die Konfidenz einer Diagnose, erklärte Oqueka weiter.8 Allerdings liefert sie nur wenige konkrete Diagnosen und auch die Standardisierung muss noch weiter vorangetrieben werden. Konkret diagnostizieren lassen sich mittels BAL beispielsweise das bronchoalveoläre Karzinom (Tumorzellen), die eosinophile Pneumonie (hohe Eosinophilenzahl) oder eine diffuse alveoläre Hämorrhagie.9
BAL und Milchglastrübungen
Der Nachweis von Entzündungszellen in der BAL korreliert nicht mit Milchglastrübungen, da diese nicht zwangsläufig Auffälligkeiten wie Lymphozyten oder Entzündungszellen in der BAL zur Folge haben, sodass die BAL hier – auch verbunden mit einem CT-Bild – wenig hilfreich ist.10
BAL und Lymphozytose
Zur Bedeutung und Häufigkeit einer Lymphozytose habe sich feststellen lassen, dass bei ausgeprägter Fibrose (≥20%) oder Vorliegen eines UIP-Musters in der Regel selten eine Lymphozytose vorliege, so Oqueka. Bei gering ausgeprägter Fibrose und Fehlen des UIP-Musters geht eine hohe Lymphozytose eher mit einem besseren Verlauf einher. Möglicherweise eignet sich also die Lymphozytose als Marker für den Verlauf dieser ILD.11
BAL und Neutrophile
Bei SSc eignen sich neutrophile Granulozyten in der BAL nicht als Marker für den Verlauf einer Lungenfibrose. Zwar ist bei höherer Neutrophilenzahl das Ausmaß der Fibrose größer und die Lungenfunktion schlechter, doch die Neutrophilenzahl alleine hat keine Aussagekraft hinsichtlich Therapieansprechen, FVC-Verlust, progressionsfreien Überlebens oder Spätmortalität.12, 13
Therapieoptionen bei SSc- und RA-assoziierter IDL
Eine SSc-assoziierte Lungenbeteiligung weist häufig ein NSIP-Muster auf, eine RA-assoziierte eher ein UIP-Muster. Die wissenschaftliche Evidenz hinsichtlich der Therapieoptionen beschrieb Oqueka folgendermaßen: Bei der SSc könne man eine therapeutische Immunmodulation mit Cyclophosphamid (Cyc), Mycophenolat Mofetil (MMF), Tocilizumab und Rituximab vornehmen. Für eine antifibrotische Therapie stehe Nintedanib zur Verfügung, das den FVC-Verlust verlangsamen könne, also das Korrelat für ein Fortschreiten der Fibrose. Die Kombination aus beiden Therapien, vor allem aus MMF und Nintedanib, sei offenbar vorteilhaft. Dass dies auch grundsätzlich für die Kombination aus antiinflammatorischer und antifibrotischer Therapie gelten könnte, wäre plausibel, da eine NSIP sehr häufig auch eine entzündliche Komponente habe.14-15
Die ILD bei RA weist ein UIP-Muster auf. Die Datenlage hinsichtlich ihrer antiinflammatorischen Therapie ist schlechter. Eine aktuelle Analyse von RABBIT-Register-Daten hat jedoch gezeigt, dass Menschen mit RA, die keine Therapie mit „dis-ease-modifying antirheumatic drugs“ (DMARDs) erhalten, ein doppelt so hohes Mortalitätsrisiko tragen wie jene, die eine solche Therapie erhalten hatten. Die Mortalität lag im Mittel bei 35% während einer Beobachtungsdauer von gut zwei Jahren.
Die Daten aus dem RABBIT-Register stammen aus der langfristigen Beobachtung der RA-Therapie mit Biologika, Biosimilars und Januskinase(JAK)-Inhibitoren bei Erwachsenen. Das Autorenteam der Studie ging davon aus, dass biologische Non-TNFi-DMARDs einen gesteigerten Nutzen bei RA-ILD haben können.17 Die RA-ILD solle aufgrund dieser – bisher nur retrospektiven – Daten antiinflammatorisch therapiert werden, empfahl Oqueka. Hierfür spreche auch, dass RA und SSc bei starker systemischer Entzündung schwerer verliefen und die Mortalität bei SSc-ILD zurückgehe, wenn die Inflammation therapeutisch reduziert werden könne. Patientinnen und Patienten mit progressiver RA-ILD profitieren zudem ebenso von einer antifibrotischen Therapie mit Nintedanib wie jene mit idiopathischer oder SSc-assoziierter Lungenfibrose.18
Von einer immunsuppressiven Behandlung mit Prednison, Azathioprin und N-Acetylcystein der IPF ist man hingegen abgerückt, da sie mit einer erhöhten Mortalität und Hospitalisierungsrate verknüpft war.19 Wie die IPF hat die RA-ILD ein UIP-Muster und inflammatorische Aspekte und ähnelt deren Verlauf. Die Frage, ob RA-ILD-Betroffene von einer Immunsuppression profitieren, sei noch nicht abschließend zu beantworten.
Insgesamt müsse man immer zunächst entscheiden, ob inflammatorische oder progressiv fibrotische Aspekte eine Erkrankung dominieren oder ein Zustand dazwischen vorliegt. Ein Teil der Patientinnen und Patienten profitiere von einer kombinierten antiinflammatorischen und antifibrotischen Therapie sehr, darauf wiesen die aktuellen Daten hin, schloss Oqueka.
Quelle:
„Pulmonale Inflammation vs. Fibrose – vom Entzündungsmuster zur Therapiekonsequenz“, Vortrag von Dr. Tim Oqueka, Hamburg, anlässlich des DGRh 2024
Literatur:
1 Arakawa H et al.: AJR Am J Roentgenol 2011; 196: 773-872 2 Wijsenbeek M et al.: N Engl J Med 2020; 383: 958-68 3 Behr J et al.: Pneumologie 2023; 77: 94-119 4 Smith ML et al.: Mod Pathol 2021; doi:10.1038/s41379-021-0889-5 5 Moran-Mendoza O et al.: BMJ Open Resp Res 2021; doi:10.1136/bmjresp-2020-000815 6 Criée C-P et al.: Pneumologie 2015; 69: 147-64 7 Müller-Mang C et al.: Radiographics 2007; 27: 595-615 8 Kreuter M et al.: Pneumologie 2023; 77: 269-302 9 Meyer KC et al.: Am J Crit Care 2012; 185: 1004-14 10 Grant-Orser A et al.: Chest 2025; 167: 172-82 11 Barnett JL et al.: Am J Respir Crit Care Med 2023; 208: 975-82 12 Goh NSL et al.: Arthritis Rheum 2007; 56: 2005-12 13 Strange C et al.: Am J Respir Crit Care Med 2008; 177: 91-8 14 Tashkin DP et al.: Lancet Respir Med 2016; 4: 708-19 15 Khanna D et al.: Lancet Respir Med 2020; 8: 963-74 16 Distler O et al.: N Engl J Med 2019; 380: 2518-18 17 Rudi T et al.: RMD Open 2024; doi 10.1136/rmdopen-2023-003789 18 Flaherty KR et al.: N Engl J Med 2019; 381: 1718-27 19 Idiopathic Pulmonary Fibrosis Clinical Research Network. N Engl J Med 2012; 366: 1968-1977 20 Raghu G et al.: Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205(9): e18-e47
Das könnte Sie auch interessieren:
Neue Therapieansätze für Arthrose
Dass Zellen altern, könnte eine entscheidende Rolle bei der Entstehung von Arthrose spielen. Welche Mechanismen dahinterstecken und welche Ansätze sich für neue Therapien ergeben, ...

Fertilität und Schwangerschaft bei entzündlicher Arthritis
Auf der 13. International Conference on Reproduction, Pregnancy and Rheumatic Diseases (RheumaPreg 2025) in Wien präsentierte Prof. Dr. Radboud Dolhain (Rotterdam, NL) aktuelle ...
So beeinflusst die Menopause rheumatische Erkrankungen
Die Menopause stellt für Patientinnen mit rheumatischen Erkrankungen eine besondere Herausforderung dar. Durch den Östrogenmangel kommt es nicht nur zu typischen Wechseljahresbeschwerden ...