Fokus rechtes Herz
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Bei der jährlich wiederkehrenden Veranstaltung „Fokus rechtes Herz“ diskutierten drei Topexperten aus Wien anhand von zwei Fällen Diagnostik, Differenzialdiagnosen und Zusammenhänge zwischen Herz und Lunge. Klar ist: Sobald eines der beiden Organe erkrankt, leidet meist auch das andere.
Wir machen diese Veranstaltung zum Thema rechtes Herz mit Janssen schon seit ungefähr zwölf Jahren, und das ist eine echte Erfolgsgeschichte“, sagte Univ.-Prof. Dr. Irene Lang, Universitätsklinik für Innere Medizin II, MedUni Wien, einleitend. Allerdings war das rechte Herz sozusagen nur der Aufhänger, anhand dessen sich komplexe Falldiskussionen entspannen, die bis weit in andere Gebiete der Medizin hineinreichten.
Was hat diese Patientin?
„Es handelt sich hier um eine 56-jährige Patientin, 173cm groß, 70kg schwer, BMI 25,3kg/m2, RR 96/61mmHg. Sie raucht drei bis fünf Zigaretten pro Tag und war immer sportlich. Im September 2021 erlitt sie plötzlich, von einem Tag auf den anderen, einen Leistungsknick, konnte nicht mehr laufen, ja bald auch nicht einmal mehr gehen“, schilderte Lang. „So etwas sieht man typischerweise bei PAH-Patienten; allerdings liegt bei idiopathischer PAH das Erstmanifestationsalter um die 30 Jahre, während es bei anderen PAH-Formen höher ist, wie wir aus aktuellen Registerdaten wissen.“
Die Patientin wird stationär aufgenommen. Die O2-Sättigung liegt bei 98%, sie klagt über Schmerzen am rechten Rippenbogen. Im Ruhe-EKG findet sich ein Sinusrhythmus, ein Normal- bis Steiltyp, aber keine typischen Zeichen einer pulmonalen Druckerhöhung. Allerdings ist sie mit einer Herzfrequenz von 100/min in Ruhe doch auffallend tachykard. Eine Atemwegserkrankung besteht nicht, jedoch finden sich im Thoraxröntgen beidseits Pleurawinkelergüsse, weiters besteht ein Aszites. Die Patientin hüstelt und räuspert sich beim Sprechen. In der Auskultation zeigt sich ein diskretes Knisterrasseln. Das Blutbild bietet eine makrozytäre Anämie mit einem Hb von 9,2g/dl und eine leichte Thrombopenie mit 136G/l. Die Leukozyten sind im unteren Normalbereich. Es findet sich kein Hinweis auf eine Blutungsquelle. Kolo- und Gastroskopie sind unauffällig.
„An dieser Stelle zogen wir folgende Differenzialdiagnosen in Betracht: Linksherzinsuffizienz mit erhaltener Auswurffraktion (HFpEF), akute Pulmonalembolie (PE), Abriss eines Trikuspidalsegels, Vorhofseptumdefekt oder auch Malignom“, berichtete die Kardiologin.
Weitere Laborwerte: Das Kreatinin (1,42mg/dl), die Gamma-GT (72U/l) und die LDH (282U/l) sind erhöht, das proBNP (3095pg/ml) ist stark erhöht, das CRP im Normalbereich. Das Gesamteiweiß ist reduziert, die Elektrophorese jedoch weitgehend normal. Ein Alkoholproblem besteht mit Sicherheit nicht. Somit ist klar, dass eine Herzinsuffizienz (HI) vorliegt, die Ursache muss aber noch gefunden werden.
Sämtliche Antikörper, die auf eine Bindegewebserkrankung hinweisen könnten, sind negativ. Allerdings zeigen sowohl eine kardiale MRT als auch eine Echokardiografie einen dilatierten rechten Vorhof und Ventrikel (jedoch keine Wandhypertrophie) und eine ausgeprägte Trikuspidalinsuffizienz (TI). Auch der linke Ventrikel erscheint vergrößert.
Die Diagnose lautet also mittel- bis höhergradige TI. Dies war auch auskultatorisch hörbar, es fand sich ein weiches systolisches Geräusch. Die HF ist weiterhin mehr oder weniger konstant um die 100–110/min.
Trikuspidalklappe: Täter oder Opfer?
„An dieser Stelle kann man sich zwei Fragen stellen“, erklärte Univ.-Prof. Dr. Thomas Binder, Universitätsklinik für Innere Medizin II, MedUni Wien. „Die eine Frage lautet, warum die Patientin eine TI hat, die andere, ob die TI die Vergrößerung des rechten Herzens und den Pleuraerguss erklärt.“
Zunächst ist zwischen strukturellen und funktionellen Ursachen einer TI zu unterscheiden. Mögliche Ursachen für eine strukturelle TI sind rheumatische Erkrankungen, Trauma, Prolaps, Endokarditis, Karzinoid oder kongenitale Fehlbildungen. Bei Weitem häufiger sind aber funktionelle Ursachen, von denen es eine ganze Reihe gibt. Im Prinzip beruhen sie auf einem von zwei Mechanismen (oder auch auf beiden): einerseits der Dilatation des Geweberings (Anulus), der die Klappe außen begrenzt, andererseits auf dem sogenannten Tethering (Zug auf die Klappensegel). Dies kann z.B. bei pulmonaler Hypertonie (PH) der Fall sein (wobei das Ausmaß der pulmonalarteriellen Druckerhöhung noch nichts über das Ausmaß der TI aussagt). „In dieser Situation ist die nichtinvasive Bestimmung des pulmonalarteriellen Drucks problematisch“, sagte Binder. „Die rechtsventrikuläre Funktion wird überschätzt, weil die TI die Nachlast reduziert; der Cardiac Output fällt ab. Diese Situation ist jedoch reversibel.“
Andere mögliche Ursachen sind Erkrankungen der Mitral- oder Aortenklappe. Auch nach Klappenersatz kann es zu einer TI kommen. Linksventrikuläre Dysfunktion (auch HFpEF) kann ebenfalls zu einer TI führen, ebenso auch eine Dysfunktion des rechten Ventrikels. „Bei dilatativer Kardiomyopathie ist die TI ein Prognosefaktor und dient als Indikator für eine PH; die TI kann auch als Verlaufsparameter für die Therapie betrachtet werden“, erklärte Binder.
Ein weiterer möglicher Grund für eine TI ist Vorhofflimmern (VHF). Dieses ist eine Ursache für eine Dilatation der Vorhöfe. „Schon aus diesem Grund war ich immer der Meinung, dass die Rhythmuskontrolle bei VHF der Frequenzkontrolle vorzuziehen ist, denn diese Vorhofdilatation ist reversibel, wenn wieder ein Sinusrhythmus einsetzt“, fuhr der Kardiologe fort. „Was uns die Erfahrung lehrt, ist, dass eine Rechtsherzinsuffizienz nicht allein durch eine TI entsteht, sondern nur dann, wenn der Ventrikel selbst ein funktionelles Problem hat“, erklärte Binder.
Die nichtinvasive, d.h. echokardiografische Abschätzung des Drucks im pulmonalarteriellen System ist bei der konkreten Patientin schon deshalb kaum möglich, weil sie aufgrund der TI keinen Druckgradienten zwischen rechtem Vorhof und rechtem Ventrikel mehr aufweist. Es findet sich aber ein turbulenter Fluss in der Pulmonalarterie. Die Volumsmessung ergibt ein Volumen von 138ml für den linken Ventrikel, das entspricht einer eindeutigen Dilatation. Und auch der rechte Vorhof und der rechte Ventrikel sind klar dilatiert. Der Fluss durch die Aortenklappe ist erhöht. „Das sieht also aus wie eine Volumenbelastung“, kommentierte Binder. Ein Vorhofseptumdefekt wird durch eine Kontrastmitteluntersuchung ausgeschlossen.
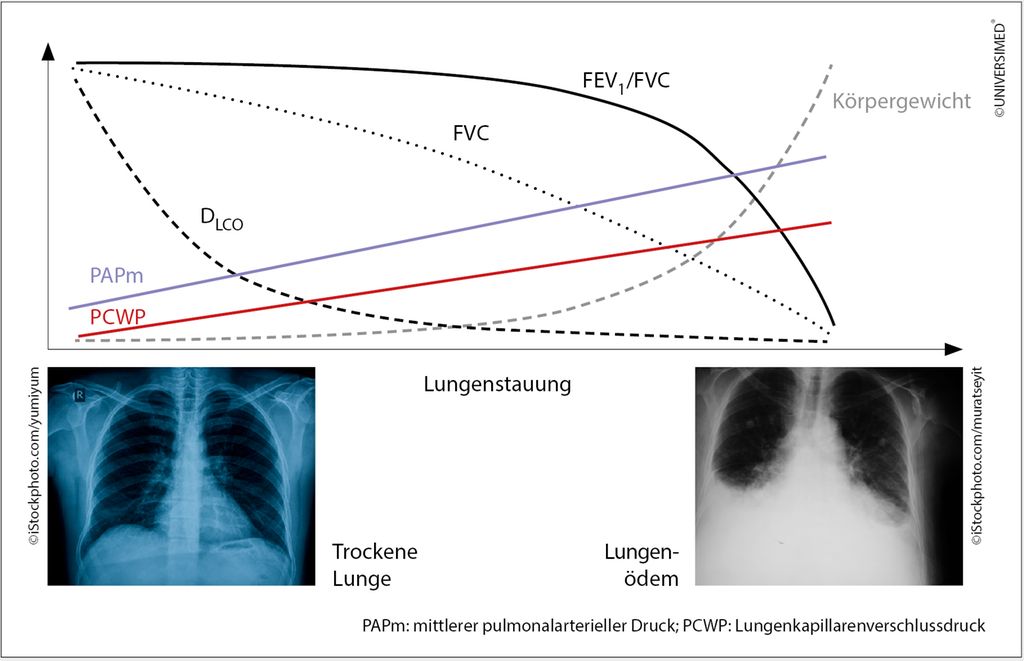
Abb. 1: Lungenfunktion und HI (modifiziert nach Magnussen H et al.: Eur J Heart Fail 2017; 9(10): 1222-9)
Des Rätsels Lösung
„Der nächste Schritt in der Abklärung war der Rechtsherzkatheter“, übernahm nun wieder Lang. Es fand sich ein pulmonalarterieller Druck von 38/18/27mmHg (systolisch/diastolisch/Mitteldruck) und somit eine PH. Der Wedgedruck war mit 21mmHg ebenfalls erhöht (Grenzwert sind 15mmHg). „Somit bestand eine postkapilläre Form der pulmonalarteriellen Druckerhöhung. Wir fanden eine normale Sauerstoffsättigung, aber ein erhöhtes Herzzeitvolumen. Damit können wir von einem ,High Output Heart Failure‘ sprechen“, berichtete die Kardiologin. In einer ebenfalls durchgeführten Koronarangiografie zeigte sich kein Hinweis auf ein pathologisches Geschehen in den Herzkranzgefäßen.
Für einen High Output Heart Failure gibt es im Wesentlichen fünf Ursachen: Adipositas, Lebererkrankung, arteriovenöse Fisteln, Lungenerkrankung und myeloproliferative Erkrankungen. „Zunächst schienen alle diese Ursachen auszuscheiden“, so Lang weiter. „Die Elektrophorese war normal, wir fanden dann jedoch eine erhöhte Anzahl von Plasmazellen im Blutausstrich und in der Folge auch im Knochenmark. Die Diagnose lautete schließlich: Kappa-Leichtketten-Myelom.“
Die Patientin besserte sich auf eine Therapie mit Bortezomib. Die Prognose ist mittelfristig jedoch leider schlecht. „Der genaue Mechanismus, durch den ein myeloproliferatives Syndrom zu einem hyperdynamen Kreislaufgeschehen führt, ist übrigens nicht geklärt“, so Lang abschließend. „Mitnehmen kann man sicher, dass jeder echokardiografische Befund letztlich eine Erklärung finden muss.“
COPD oder Herzinsuffizienz?
„Ich denke immer wieder über folgenden Fall nach und lerne davon“, erzählte Prim. Univ.-Prof. Dr. Georg-Christian Funk, 2. Medizinische Abteilung mit Pneumologie, Klinik Ottakring, Wien. Der 77-jährige Mann hat eine arterielle Hypertonie, einen Typ-2-Diabetes und ein paroxysmales Vorhofflimmern. Aufgenommen wird er wegen Belastungsdyspnoe. Im EKG zeigt er einen Linkstyp, einen Rechtsschenkelblock und einen linksanterioren Hemiblock. Das NT-proBNP liegt bei 1402pg/ml. In der Lungenfunktion zeigen sich eine nicht reversible Obstruktion und eine Restriktion, das Residualvolumen ist nicht vergrößert. „Wir gingen von einer COPD aus, die auch anamnestisch beschrieben war“, so Funk.
Der Patient wurde mit Betablockern und ACE-Hemmern behandelt, die Herzinsuffizienz besserte sich dramatisch. „Und bei einer Lungenfunktionskontrolle nach drei Monaten hatte sich die angebliche COPD in Luft aufgelöst, es bestand keine Obstruktion mehr. Dies trifft auf etwa die Hälfte aller Patienten zu, bei denen in der Herzinsuffizienz eine COPD diagnostiziert wird. Das ist etwas, was man gar nicht machen sollte“, mahnte Funk. Abbildung 1 zeigt die Zusammenhänge zwischen zunehmender Lungenstauung aufgrund der HI und einigen wichtigen Parametern der Lungenfunktion.
Fazit
Die vorgestellten Fälle haben einmal mehr verdeutlicht, wie komplex sich die Diagnosestellung gestalten kann, wenn Herz und Lunge involviert sind. Umso wichtiger für die optimale Betreuung betroffener Patienten ist daher die enge Kooperation – sowohl zwischen den unterschiedlichen Disziplinen als auch auf den verschiedenen Ebenen des Gesundheitssystems.
Entgeltliche Einschaltung
Mit freundlicher Unterstützung durch Janssen-Cilag Pharma GmbH
AT_ CP-276765_01Feb2022
Quelle
Webinar „Fokus rechtes Herz“ von Janssen-Cilag Pharma GmbH am 18.11.2021
Literatur:
bei den Vortragenden
Fachkurzinformation Uptravi®
Bezeichnung des Arzneimittels: Uptravi 200µg / 400µg / 600µg / 800µg / 1000µg / 1200µg / 1400µg / 1600µg Filmtabletten. Qualitative und quantitative Zusammensetzung: Eine Filmtablette (FT) enthält entweder 200µg, 400µg, 600µg, 800µg, 1000µg, 1200µg, 1400µg oder 1600µg Selexipag. Sonstige Bestandteile: Tablettenkern: Mannitol (E421), Maisstärke, niedrig substituierte Hyprolose, Hydroxypropylcellulose, Magnesiumstearat. Filmüberzug: Hypromellose, Propylenglycol, Titandioxid (E171), Carnaubawachs und Eisenoxide (Uptravi 200µg FT: Eisen(III)-hydroxid-oxid x H2O (E172); Uptravi 400µg FT: Eisen(III)-oxid (E172); Uptravi 600µg FT: Eisen(III)-oxid (E172) und Eisen(II,III)-oxid (E172); Uptravi 800µg FT: Eisen(III)-hydroxid-oxid x H2O (E172) und Eisen(II,III)-oxid (E172); Uptravi 1000µg FT: Eisen(III)-oxid (E172) und Eisen(III)-hydroxid-oxid x H2O (E172); Uptravi 1200µg FT: Eisen(II,III)-oxid (E172) und Eisen(III)-oxid (E172); Uptravi 1400µg FT: Eisen(III)-hydroxid-oxid x H2O (E172); Uptravi 1600µg FT: Eisen(II,III)-oxid (E172), Eisen(III)-oxid (E172) und Eisen(III)-hydroxid-oxid x H2O (E172)). Anwendungsgebiet: Uptravi ist indiziert für die Langzeitbehandlung der pulmonal arteriellen Hypertonie (PAH) bei erwachsenen Patienten der WHO-Funktionsklasse (WHO-FC) II bis III entweder als Kombinationstherapie bei Patienten, deren Erkrankung mit einem Endothelin-Rezeptor-Antagonisten (ERA) und/oder einem Phosphodiesterase-5(PDE-5)-Inhibitor unzureichend kontrolliert ist oder als Monotherapie bei Patienten, die für diese Therapien nicht infrage kommen. Die Wirksamkeit wurde bei PAH, einschließlich idiopathischer und erblicher PAH, PAH in Assoziation mit Bindegewebserkrankungen und PAH in Assoziation mit korrigierten einfachen angeborenen Herzfehlern nachgewiesen. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile; schwere koronare Herzerkrankung oder instabile Angina pectoris; Myokardinfarkt innerhalb der letzten 6 Monate; dekompensierte Herzinsuffizienz, sofern nicht engmaschig überwacht; schwere Arrhythmien; zerebrovaskuläre Ereignisse (z.B. transiente ischämische Attacke, Schlaganfall) innerhalb der letzten 3 Monate; angeborene oder erworbene Klappendefekte mit klinisch relevanten myokardialen Funktionsstörungen, die nicht mit einer pulmonalen Hypertonie in Verbindung stehen; gleichzeitige Anwendung von starken CYP2C8-Inhibitoren (z.B. Gemfibrozil). Inhaber der Zulassung: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgien. Vertrieb in Österreich: JANSSEN-CILAG Pharma GmbH, Vorgartenstraße 206B, A-1020 Wien. Verschreibungspflicht/Apothekenpflicht: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. ATC-Code: B01AC27. Weitere Angaben zu Warnhinweisen und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln und sonstigen Wechselwirkungen, Schwangerschaft und Stillzeit, sowie Nebenwirkungen entnehmen Sie bitte der veröffentlichten Fachinformation. Uptravi_FKI_CP-201712_20210326
Fachkurzinformation OPSUMIT®
Bezeichnung des Arzneimittels: Opsumit 10 mg Filmtabletten. Qualitative und quantitative Zusammensetzung: Jede Filmtablette enthält 10 mg Macitentan. Sonstige Bestandteile mit bekannter Wirkung: jede Filmtablette enthält ungefähr 37 mg Lactose (als Monohydrat) und ungefähr 0,06 mg Phospholipide aus Sojabohnen (E322). Sonstige Bestandteile: Tablettenkern: Lactose-Monohydrat, mikrokristalline Cellulose (E460i), Carboxymethylstärke-Natrium (Typ A), Povidon K-30, Magnesiumstearat (E572), Polysorbat 80 (E433). Filmüberzug: Poly(vinylalkohol) (E1203), Titandioxid (E171), Talkum (E533b), Phospholipide aus Sojabohnen (E322), Xanthangummi (E415). Anwendungsgebiet: Opsumit, als Monotherapie oder in Kombination, ist indiziert für die Langzeitbehandlung der pulmonal-arteriellen Hypertonie (PAH) bei erwachsenen Patienten mit WHO-Funktionsklasse (WHO-FC) II bis III. Die Wirksamkeit wurde bei Patienten mit PAH nachgewiesen einschließlich idiopathischer und erblicher PAH, PAH in Assoziation mit Bindegewebserkrankungen sowie PAH in Assoziation mit korrigierten einfachen angeborenen Herzfehlern. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff, Soja oder einen der sonstigen Bestandteile; Schwangerschaft; Frauen im gebärfähigen Alter, die keine zuverlässigen Verhütungsmethoden anwenden; Stillzeit; Patienten mit schwerer Leberfunktionsstörung (mit oder ohne Zirrhose); vor Behandlungsbeginn bestehende Erhöhung der Leber-Aminotransferasewerte (Aspartat-Aminotransferase (AST) und/oder Alanin-Aminotransferase (ALT)) >3 x ONW. Inhaber der Zulassung: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgien. Vertrieb in Österreich: JANSSEN-CILAG Pharma GmbH, Vorgartenstraße 206B, A-1020 Wien. Verschreibungspflicht/Apothekenpflicht: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. ATC-Code: C02KX04. Weitere Angaben zu Warnhinweisen und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen, Schwangerschaft und Stillzeit, sowie Nebenwirkungen entnehmen Sie bitte der veröffentlichten Fachinformation. AT_CP-158604_20210520