MME-Sequencing bei „late-onset“ hereditären Neuropathien
Hereditäre Polyneuropathien, auch bekannt als Charcot-Marie-Tooth-Syndrom (CMT), sind bereits seit dem Ende des 19. Jahrhunderts bekannt und erfordern sehr häufig eine kontinuierliche neurologische und orthopädische Betreuung. Während die Erstdiagnose bisher überwiegend bei Kindern, Jugendlichen und im frühen Erwachsenenalter gestellt wurde, haben klinische und genetische Untersuchungen im letzten Jahrzehnt ganz klar gezeigt, dass der Krankheitsbeginn in vielen Fällen auch erst wesentlich später sein kann. Liegt er nach dem 35. Lebensjahr oder auch deutlich danach, spricht man von „late-onset hereditären Neuropathien“ (LOCMT).1–3 Diese sind mittlerweile eine wichtige Differenzialdiagnose der häufigen chronisch-idiopathischen Polyneuropathien des Alters geworden.4,5
Was sind die typischen Symptome der LOCMT?
Nicht selten berichten die Patienten initial über Parästhesien bzw. eine Hypästhesie in den Zehen bzw. treten die sensiblen Ausfälle manchmal auch isoliert im Bereich der Großzehe ein- oder beidseitig auf und breiten sich allmählich bis zum Vorfuß und dann weiter nach proximal aus. Sie können auch mit neuropathischen Schmerzen unterschiedlicher Intensität einhergehen. Je nach genetischer Ursache treten auch distale motorische Ausfälle an den unteren Extremitäten auf, die zunächst als eingeschränkte Beweglichkeit der Zehen und in weiterer Folge als Fußheberschwäche wahrgenommen werden. Das Gangbild verändert sich, der Gang wird unsicher, es besteht eine Fallneigung. Im Unterschied zu den klassischen hereditären Neuropathien sind die Hände nicht bzw. wesentlich später und meist nur in geringem Ausmaß betroffen.1–3
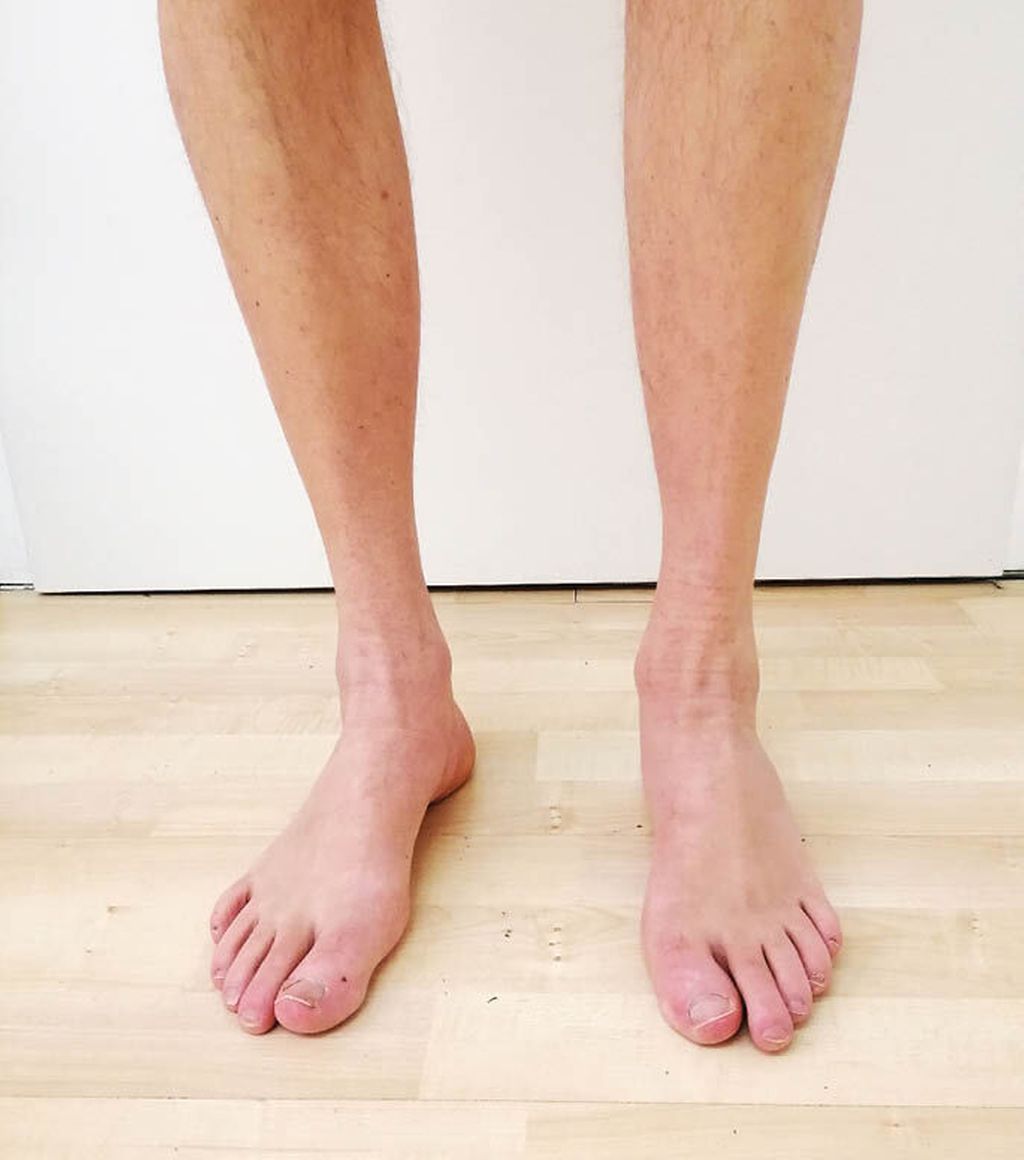
Abb. 1: Distal betonte Atrophie der Unterschenkelmuskulatur sowie Hohlfußbildung bei einem Patienten mit MME-Mutation
Die neuroorthopädische Untersuchung zeigt eine Atrophie der kleinen Fußmuskeln mit Hammerzehen- und (meist nur geringer bis mäßiger) Hohlfußbildung bds., sowie je nach Krankheitsstadium eine variable Ausprägung der sensiblen und motorischen Ausfälle mit einer oft sehr ausgeprägten Atrophie der Wadenmuskulatur bds. (Abb. 1). Der Gang entspricht dem typischen Steppergang, jedoch besteht fast immer auch eine erhebliche Ataxie, die der Patient meist explizit anamnestisch als „Gleichgewichtsstörung“ hervorhebt. Diese fällt insbesondere durch ein starkes Schwanken nach dem Aufstehen und im Stehen auf, in fortgeschrittenen Krankheitsstadien muss sich der Patient daher z.B. an einer Tischkante festhalten, um nicht zu stürzen. Andere Koordinationsstörungen, wie z.B eine Ataxie im Knie-Hacken-Versuch, sind nicht typisch. Die Muskeleigenreflexe an den unteren Extremitäten sind meist abgeschwächt oder fehlen.
Die Untersuchung der Nervenleitgeschwindigkeit (NLG) ergibt eine primär axonale Schädigung der motorischen und sensiblen Nerven.
LOCMT und Familienanamnese
Besteht der Verdacht auf eine genetische Ursache der Polyneuropathie, so ist die Erhebung einer ausführlichen Familienanamnese mit Befragung nach ähnlichen Symptomen bei den Eltern und Geschwistern bzw. weiteren Verwandten erforderlich. Aufgrund des oft sehr späten Krankheitsbeginns lässt sich oft kein Hinweis für eine familiäre Häufung feststellen, da in vielen Fällen die Erkrankung zum Zeitpunkt des Todes des betroffenen Elternteils noch nicht ausgebrochen war. Eine negative Familienanamnese schließt daher das Vorliegen einer LOCMT keineswegs aus!
Die wichtigsten Differenzialdiagnosen
Die in höherem Lebensalter besonders häufigen diabetischen Neuropathien sind meist aufgrund des wesentlich langsameren Verlaufs gut abgrenzbar. Auch stehen bei diabetischen Neuropathien motorische Ausfälle meist im Hintergrund. Selbstverständlich können aufgrund der hohen Prävalenz der diabetischen Neuropathien diese auch gleichzeitig mit einer LOCMT vorkommen.3
Alkoholische Neuropathien lassen sich meist anamnestisch ausschließen bzw. auch durch Fehlen anderer Folgeerscheinungen des erhöhten Alkoholkonsums gut von der LOCMT abgrenzen.
Bei immunmediierten chronisch-idiopathischen demyelinisierenden Polyneuropathien (CIDP) kann der klinische Verlauf sehr ähnlich sein, jedoch ist eine Abgrenzung aufgrund typischer NLG-Veränderungen bei CIDP meist problemlos möglich.
Weiters sind bei unklarer Ursache einer progredienten Polyneuropathie des höheren Lebensalters paraneoplastische Formen auszuschließen.
Dennoch bleibt trotz umfassender Durchuntersuchung die Ursache oft ungeklärt. Man spricht dann von der chronisch-idiopathischen Polyneuropathie des höheren Lebensalters.4,5 Gerade in diesen Fällen wird die weitere genetische Abklärung empfohlen.
Mutationen im MME-Gen stellen die häufigste Ursache der LOCMT dar
Im Jahr 2017 wurden unabhängig voneinander von drei Arbeitsgruppen Mutationen im MME-Gen, welches das Enzym Neprilysin kodiert, beschrieben. Während ein japanisches Forschungsteam ausschließlich Patienten mit rezessiver Vererbung beschrieb,1 wurde andererseits auch über einige Familien berichtet, in denen bereits heterozygote Mutationen im MME-Gen zum typischen Phänotyp der LOCMT führen.2 Neben den meist deutlichen rasch progredienten motorischen Ausfällen wurde die zusätzliche ataktische Komponente von einer weiteren Arbeitsgruppe besonders hervorgehoben und die Erkrankung auch als spinozerebelläre Ataxie klassifiziert.6
2020 wurde eine sehr große internationale Follow-up-Studie in der Zeitschrift „Neurology“ veröffentlicht, bei der 230 Patienten mit unklarer axonaler Polyneuropathie und Krankheitsbeginn nach dem 35. Lebensjahr genetisch untersucht wurden. Dabei konnte bei knapp 1/5 aller Patienten eine eindeutige genetische Ursache detektiert werden. Bei Patienten mit positiver Familienanamnese konnte die zugrunde liegende Mutation sogar bei mehr als 1/4 aller Patienten festgestellt werden. Dabei waren Mutationen im MME-Gen die bei Weitem häufigste Ursache.3
Die für MME-Mutationen besonders typischen Krankheitszeichen und die am häufigsten betroffenen Gene sind in den Textkästen unten aufgelistet.
Orthopädische Versorgung von Patienten mit LOCMT
Bei der LOCMT ist die möglichst frühe Diagnose von besonderer Bedeutung. Entscheidend ist die optimale Stabilisierung des Gangbildes zur Verbesserung der Gehfähigkeit und zur Vermeidung von Stürzen. Die entsprechenden Gehhilfen müssen im Krankheitsverlauf immer wieder individuell angepasst werden. Während zunächst oft eine entsprechende Einlagenversorgung ausreichend ist, wird bei zunehmender distaler Muskelschwäche meist eine Versorgung mit Orthesen erforderlich. Eine Observanz in Abständen von 6–9 Monaten wird aufgrund der oft raschen Progredienz empfohlen. Im Gegensatz zu den hereditären Polyneuropathien mit frühem Krankheitsbeginn kommen operative Verfahren bei der LOCMT kaum zum Einsatz. Die Therapie der oft begleitenden neuropathischen Schmerzen sollte in Absprache mit dem behandelnden Neurologen erfolgen.
Literatur:
1 Higuchi Y et al.: Mutations in MME cause an autosomal- recessive Charcot-Marie-Tooth disease type 2. Ann Neurol 2016; 79: 659-72 2 Auer-Grumbach M et al.: Rare variants in MME, encoding metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am J Hum Genet 2016; 99: 607-23 3 Senderek J et al.: The genetic landscape of axonal neuropathies in the middle-aged and elderly: focus on MME. Neurology 2020; 95: e3163-7 4 Zis P et al.: Chronic idiopathic axonal polyneuropathy: a systematic review. J Neurol 2016; 263:1903-10 5 Hanewinckel R et al.: Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016; 87: 1892-8 6 Depondt C et al.: MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43). Neurol Genet 2016; 2: e94
Das könnte Sie auch interessieren:

«Auch Patienten mit Demenz profitieren von einer chirurgischen Stabilisierung»
Patienten mit Hüftfraktur und einer leichten, mittelschweren oder schweren Demenz haben ein geringeres Risiko zu sterben, wenn sie operiert werden – vor allem wenn es sich um Kopf-Hals- ...
Stellenwert des individuellen Alignments in der Knieendoprothetik
Dieser Artikel erläutert die unterschiedlichen Alignmentkonzepte in der Knieendoprothetik und deren Stellenwert. Es ist davon auszugehen, dass die Rekonstruktion der individuellen ...

Patientenoptimierung in der orthopädischen Chirurgie
Die Patientenoptimierung vor orthopädischen Eingriffen, insbesondere in der Endoprothetik, spielt eine entscheidende Rolle für den Erfolg der Operation und die Zufriedenheit der ...