7-Jahres-Daten bestätigen Effektivität der Ibrutinib-Erstlinientherapie
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Ibrutinib ist als Einzelsubstanz und/oder als Kombinationstherapie bei der Behandlung der chronischen lymphatischen Leukämie (CLL), des Mantelzelllymphoms (MCL) und des Morbus Waldenström (MW) zugelassen.1 Mit dem irreversiblen Inhibitor der Bruton-Tyrosinkinase (BTK) wurde in diversen klinischen Studien eine signifikant verbesserte Wirksamkeit gegenüber etablierten Standardtherapien erreicht. Inzwischen besteht eine langjährige Erfahrung mit dem Einsatz von Ibrutinib. Jüngst wurden die 7-Jahres-Daten der RESONATE-2-Studie bei der Jahrestagung der European Hematology Association (EHA) 2021 präsentiert.2
Für die Erstlinientherapie mit Ibrutinib bei Patienten mit CLL oder kleinzelligem lymphozytischem Lymphom (SLL) liegen mittlerweile die 7-Jahres-Daten der Phase-III-Studie RESONATE-2 mit einer medianen Nachbeobachtungszeit von 74,9 Monaten.2 In der randomisierten Studie wurde bei insgesamt 269 Patienten mit nicht behandelter CLL im Alter von ≥65 Jahren, ohne 17p-Deletion, die Therapie mit Ibrutinib bis Tumorprogress oder Unverträglichkeit gegen bis zu 12 Zyklen Chlorambucil verglichen.
Bezüglich der Effektivität zeigte sich, dass der signifikante Vorteil der Ibrutinib-Therapie mit einer 84%igen Reduktion des Risikos für einen Progress gegenüber Chlorambucil über den langen Beobachtungszeitraum erhalten blieb (HR: 0,160; 95% CI: 0,111–0,230) (Abb. 1). Nach 6,5 Jahren lebten 61% der Patienten des Ibrutinib-Arms versus 9% des Chlorambucil-Arms ohne Progress. Auch Patienten mit genomischen Hochrisikofaktoren, inklusive eines unmutierten IGHV-Status (HR: 0,109; 95% CI: 0,063–0,189) und 11q-Deletion (HR: 0,033; 95% CI: 0,010–0,107), profitierten in Subgruppenanalysen klinisch relevant von Ibrutinib.
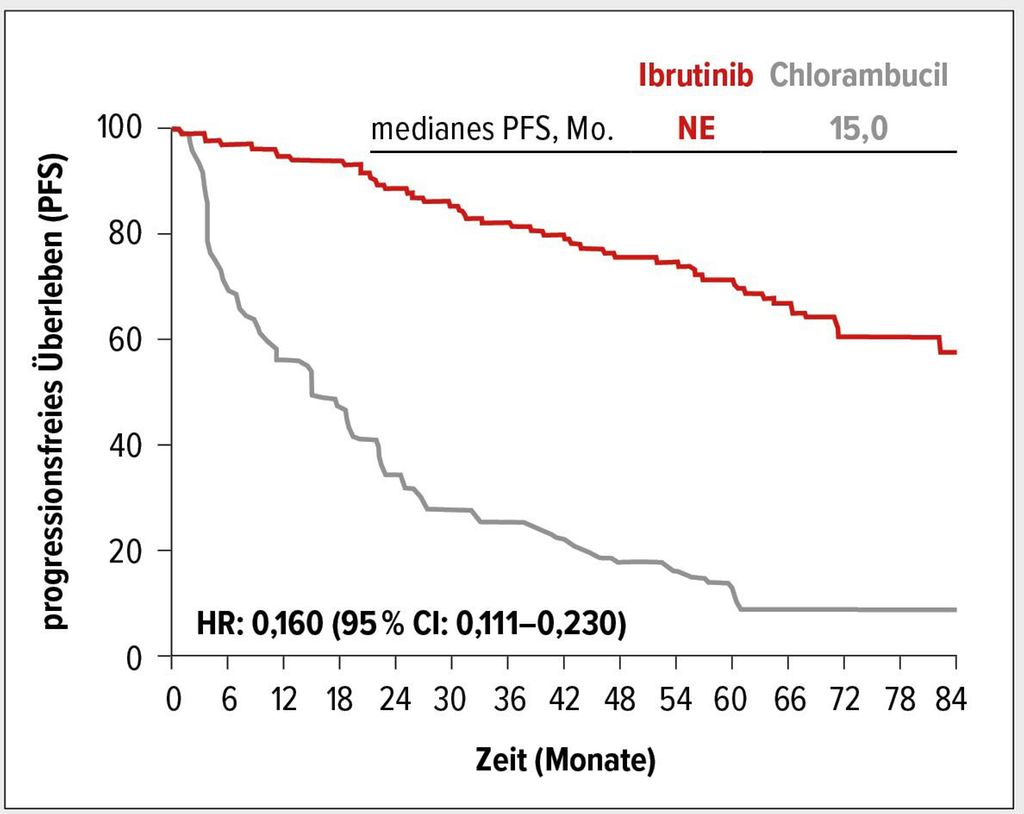
Abb. 1: Progressionsfreies Überleben (PFS) unter Ibrutinib versus Chlorambucil (mod. nach Ghia P et al.)2
Mit der Ibrutinib-Therapie wurde eine Gesamtüberlebensrate von 78% nach 6,5 Jahren erreicht. Die Ansprechrate betrug nach 6,5 Jahren 92% und die Rate an kompletten Remissionen (CR/CRi-Rate) stieg auf 34% an.
Sicherheit unter Ibrutinib langfristig gegeben
Da Ibrutinib bis zum Tumorprogress oder bis zu nicht akzeptabler Toxizität gegeben wird, hat in der Langzeitbeobachtung auch die Sicherheit einen hohen Stellenwert. Nebenwirkungen von besonderem Interesse traten selten auf: Ein Bluthochdruck Grad ≥3 wurde in den Jahren 5–6 bei 4 Patienten (5%) und in den Jahren 6–7 bei 3 Patienten (4%) beobachtet.2 Vorhofflimmern Grad ≥3 trat bei jeweils 1 Patienten (1%) in den Jahren 5–6 sowie 6–7 auf. Größere Blutungen Grad ≥3 wurden 5 bis 7 Jahre nach Therapiebeginn nicht berichtet.
Eine Dosisreduktion aufgrund von Nebenwirkungen ≥ Grad 3 wurde bei jeweils 1 Patienten im 5./6. Jahr sowie im 6./7. Jahr angegeben.2 Von 31 Patienten, bei denen irgendwann im Therapieverlauf die Dosis aufgrund einer Nebenwirkung reduziert wurde, zeigten 71% einen Rückgang oder eine Verbesserung der Nebenwirkungen. Eine Progression der CLL war in den Jahren 5–6 für 4 Patienten (5%) und in den Jahren 6–7 für 4 Patienten (6%) Grund für den Abbruch der Therapie. Insgesamt haben lediglich 16 Patienten (12%) die Therapie mit Ibrutinib in der bis zu 7 Jahre dauernden Nachbeobachtungszeit aufgrund von Progression abbrechen müssen.
Mit nunmehr 7 Jahren Nachbeobachtung verblieben 47% der Patienten unter Therapie mit der Ibrutinib-Monotherapie.
Fazit für die Praxis
Die Langzeitbeobachtung der RESONATE-2-Studie zeigt einen anhaltenden Nutzen von Ibrutinib bezüglich des progressionsfreien Überlebens (PFS) und des Gesamtüberlebens (OS) bei Erstlinienbehandlung von Patienten mit CLL/SLL. Die Remissionen vertieften sich über den Studienverlauf. Es wurden keine neuen Sicherheitssignale beobachtet und Ibrutinib wird insgesamt gut toleriert.
Entgeltliche Einschaltung
Mit freundlicher Unterstützung durch Janssen-Cilag AG
AT_CP-249267_22Jul2021
Quelle:
Jahrestagung der European Hematology Association (EHA Virtual Convention 2021), 9.–12. Juni 2021
Literatur:
1 Fachinformation Imbruvica® 2 Ghia P et al.: Ibrutinib treatment in the first-line setting for patients with chronic lymphocytic leukemia: Up to 7 years of follow-up in the RESONATE-2 study. EHA 2021, Abstr. #EP636
FKI
Bezeichnung des Arzneimittels: IMBRUVICA 140 mg Hartkapseln. IMBRUVICA 140/280/420/560 mg Filmtabletten. Qualitative und quantitative Zusammensetzung: Jede Hartkapsel enthält 140 mg Ibrutinib. Jede 140/280/420/560 mg Filmtablette enthält 140/280/420 bzw. 560 mg Ibrutinib.Sonstige Bestandteile: Croscarmellose-Natrium, hochdisperses Siliciumdioxid (alle Filmtabletten), Lactose-Monohydrat (alle Filmtabletten), Magnesiumstearat, Mikrokristalline Cellulose, Povidon (alle Filmtabletten), Natriumdodecylsulfat (E487), Gelatine (Hartkapseln), Macrogol (alle Filmtabletten), Poly(vinylalkohol) (alle Filmtabletten), Talkum (alle Filmtabletten), Titandioxid (E171), Schellack (Hartkapseln), Eisen(II,III)-oxid (E172, Hartkapseln, 140 mg, 280 mg u. 420 mg Filmtabletten), Propylenglycol (E1520, Hartkapseln), Eisen(III)-hydroxid-oxid x H2O (E172, 140 mg, 420 mg u. 560 mg Filmtabletten), Eisen(III)-hydroxid-oxid (E172, 280 mg u. 560 mg Filmtabletten).
Anwendungsgebiete: IMBRUVICA als Einzelsubstanz ist indiziert zur Behandlung erwachsener Patienten mit rezidiviertem oder refraktärem Mantelzell-Lymphom (MCL). IMBRUVICA als Einzelsubstanz oder in Kombination mit Rituximab oder Obinutuzumab ist indiziert zur Behandlung
erwachsener Patienten mit nicht vorbehandelter chronischer lymphatischer Leukämie (CLL) (siehe Abschnitt 5.1). IMBRUVICA als Einzelsubstanz oder in Kombination mit Bendamustin und Rituximab (BR) ist indiziert zur Behandlung erwachsener Patienten mit CLL, die mindestens eine vorangehende Therapie erhalten haben. IMBRUVICA als Einzelsubstanz ist indiziert zur Behandlung erwachsener Patienten mit Morbus Waldenström (MW), die mindestens eine vorangehende Therapie erhalten haben, oder zur Erstlinien-Therapie bei Patienten, die für eine Chemo-Immuntherapie nicht geeignet sind. IMBRUVICA in Kombination mit Rituximab ist indiziert zur Behandlung erwachsener Patienten mit MW.
Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Die Anwendung von Präparaten, die Johanniskraut enthalten, ist während der Therapie mit IMBRUVICA kontraindiziert. Inhaber der Zulassung: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgien. Vertrieb für Österreich: Janssen-Cilag Pharma GmbH, Vorgartenstraße 206B, A-1020 Wien. Verschreibungspflicht/Apothekenpflicht: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. ATC-Code: L01EL01. Weitere Angaben zu Warnhinweisen und
Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln und sonstigen Wechselwirkungen, Schwangerschaft und Stillzeit sowie Nebenwirkungen entnehmen Sie bitte der veröffentlichten Fachinformation.
(AT_ CP-205827_v1.0_20Jan2021)