
Lichtblicke für die Therapie von Muskelerkrankungen
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Tagung „UpDate Muskelforschung“ fand 2026 bereits zum 14. Mal statt. Trotz der Seltenheit der Muskelerkrankungen gab es in den letzten Jahren einige Fortschritte. Neben den aktuell zugelassenen Therapien wurden laufende klinische Studien und die „unmet needs“ von Patient:innen besprochen.
Die Veranstaltung „UpDate Muskelforschung“, ausgetragen von dem Verein Österreichische Muskelforschung, ist eine der führenden Tagungen für Muskelerkrankungen im deutschsprachigen Raum. Jedes Jahr treffen sich zahlreiche nationale und internationale Expert:innen anlässlich der Tagung in Wien und tauschen sich darüber hinaus mit Betroffenen und Angehörigen zu den Fortschritten und ungelösten Herausforderungen aus.
Therapien bei Muskeldystrophie Duchenne
Erwartungen, Realität und Ausblick
Die Muskeldystrophie Duchenne (DMD) ist eine X-chromosomal rezessiv vererbte Erkrankung, die durch Veränderungen im Dystrophin-Gen verursacht wird.1–3 DMD tritt bereits im frühen Alter auf, wobei sich die Symptome im Laufe der Zeit verstärken. So kommt es durch das Fehlen von Dystrophin zu einem fortschreitenden Muskelabbau und Muskelschwäche. Hinzu können zudem kognitive Beeinträchtigungen und Sprachverzögerungen kommen. Mit zunehmender Muskelschwäche sind die zumeist männlichen Patienten im Jugendalter bereits auf einen Rollstuhl angewiesen und es treten häufig respiratorische, orthopädische und kardiale Komplikationen auf.5,6
Aktuelle und neu zugelassene DMD-Therapieoptionen
„Im Mittelpunkt der DMD-Behandlung steht weiterhin das multidisziplinäre Behandlungsteam“, erklärt Dr. Anna Wiesenhofer von der Klinik Favoriten in Wien. Neben den bereits bekannten Therapien mit Prednison und Deflazacort ist seit 2/2024 auch das dissoziative Kortikosteroid Vamorolon in Österreich für Patient:innen ab 4 Jahren verfügbar.7–9 Vamorolon unterdrückt dabei die inflammatorische Reaktion, schützt die Membranstabilität und reduziert damit den Untergang der Muskelfasern bei einem geringen Nebenwirkungsprofil.8,9 Im August 2025 wurde ein Zulassungsantrag für 2- bis 4-Jährige bei der Europäischen Arzneimittelagentur (EMA) eingereicht, bisher ist der Wirkstoff nur für Patient:innen ab einem Alter von 4 Jahren zugelassen. In der Schweiz ist Vamorolon ebenso seit Anfang des Jahres 2026 zugelassen.10
Darüber hinaus wurde 6/2025 eine bedingte Zulassung für Givinostat für gehfähige DMD-Patienten ab 6 Jahren in Kombination mit einer Kortikosteroidbehandlung erteilt.11 Die Wirkung von Givinostat beruht auf der Hemmung von Histondeacetylasen (HDAC), die bei DMD überaktiv sind und folglich die regenerative Funktion der Muskelstammzellen blockieren. Zudem fördert die HDAC-Dysregulation die Entstehung chronischer Entzündungen durch Immunzellen und trägt zur Einlagerung von Fettgewebe durch fibroadipogene Vorläuferzellen und zur Fibrose bei.12 Diese Prozesse werden nachweislich durch Givinostat inhibiert und somit die Progression der DMD verzögert.13
Neue Wirkstoffe zur Erhöhung der Dystrophin-Level
Neuere Therapieansätze zielen darauf ab, die Dystrophin-Level in den Muskelzellen zu erhöhen. Die EMBARK-Studie untersuchte die Wirksamkeit des Mini-Dystrogens Delandistrogen moxeparvovec. Die 1-Jahres-Daten konnten jedoch den primären Endpunkt einer signifikanten North-Star-Ambulatory-Assessment(NSAA)-Veränderung nicht erreichen. Zwar konnten klinisch bedeutsame Verbesserungen in den sekundären Endpunkten wie der Zeit zum Aufstehen (TTR) und dem 10-Meter-Gehtest (10MWR) gezeigt werden, aber weitere Daten sind erforderlich.14 Beim Therapieansatz des Exonskippings, bei dem defekte Exons übersprungen werden und ein kürzeres, funktionales Dystrophin entsteht, wird aktuell eine neue Generation von Wirkstoffen erforscht. So wird unter anderem das Antikörper-Oligonukleotid-Konjugat AOC-1044 in einer Phase-I/II-Studie untersucht, das durch eine Anvisierung des auf Muskelzellen exprimierten Transferrin-Rezeptors eine bessere Wirkstoffaufnahme, eine gesteigerte Modifikation des Spleißens und erhöhte Dystrophin-Level verspricht.15
Spinale Muskelatrophie
Aktuelle Therapien und neue Perspektiven
Bei der spinalen Muskelatrophie (SMA) kommt es zu einer progressiven Degeneration der α-Motoneurone und einem Verkümmern der Muskulatur. Sie wird durch Deletionen im Survival-Motor-Neuron-1 (SMN1)-Gen verursacht, wobei das homologe Gen SMN2 den Verlust nur teilweise ausgleichen kann, da es nur circa 10% funktionales SMN-Protein produziert. Die Anzahl der SMN2-Kopien bestimmt somit den Phänotyp.16 „Mit Nusinersen, Risdiplam und der Gentherapie Onasemnogen-Abeparvovec stehen wirksame Behandlungsmöglichkeiten zur Verfügung“, sagt Dr. Astrid Eisenkölbl vom Kepler Universitätsklinikum in Linz: „Viele Patient:innen leiden jedoch weiterhin an Mobilitätseinschränkungen und Muskelschwäche, haben Ernährungsschwierigkeiten oder Schmerzen, was den Bedarf an verbesserten Behandlungsansätzen verdeutlicht.“17,18 Trotz immenser Fortschritte gibt es daher Bedarf an weiteren oder verbesserten Therapien und Unterstüzungsangeboten.
Vielversprechende Modifikationen bekannter SMA-Behandlungsoptionen
Ein verbesserter Behandlungsansatz ist die pränatale Therapie: In einem rezenten Fallbericht einer ungeborenen Patientin mit zwei SMN2-Kopien erhielt die Mutter ab der 32. Schwangerschaftswoche und der Säugling ab dem 8. Lebenstag Risdiplam. Eine Untersuchung im 30. Lebensmonat ergab daraufhin keine Anzeichen einer SMA.19 Aktuell wurden nun 9 weitere Fälle vorgestellt, bei denen die Säuglinge zusätzlich auch eine Gentherapie erhielten. In den meisten Fällen wurde auch hier eine unauffällige Kindesentwicklung dokumentiert.20 Dieser vielversprechende Therapieansatz bedarf nun einer näheren klinischen Begutachtung.
Anfang des Jahres wurde zudem eine Nusinersen-Hochdosistherapie von der Europäischen Komission zugelassen, nachdem die Phase-III-Studie DEVOTE positive Ergebnisse lieferte, die in Nature Medicine veröffentlicht wurden.21,22 Die zugrunde liegende Studie konnte nach 1-jähriger Nachbeobachtung bei 53% der Patient:innen eine Verbesserung des Hammersmith-Functional-Motor-Scale-Expanded (HFMSE)-Scores ermitteln. Bei 24% Patient:innen konnte keine Veränderung und bei 24% eine Verschlechterung festgestellt werden (Abb. 1).22–25 Erste Erfahrungen aus der Praxis zeigen bereits, dass die Dosisumstellung unkompliziert und die Therapie gut verträglich ist.
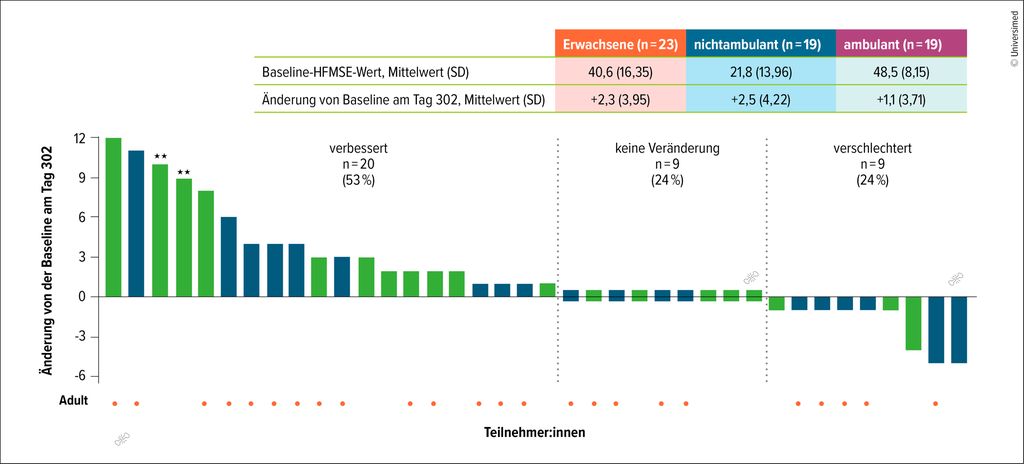
Abb. 1: Bei rund der Hälfte der Studienteilnehmer:innen mit spinaler Muskelatrophie verbesserte sich das Outcome unter Hochdosistherapie mit Nusinersen. Seit Anfang 2026 ist das Hochdosisregiment daher auch von der Europäischen Komission zugelassen. (Modifiziert nach Finkel RS et al. 202323 und Crawford T et al. WMS 202424)
SMA-Therapieansätze zur Steigerung der Erregbarkeit von Muskelzellen
Neben diesen Therapiemodifikationen werden auch neue Wirkmechanismen untersucht. So läuft derzeit eine Studie zum monoklonalen Antikörper ARGX-119, einem Agonisten der muskelassoziierten Rezeptor-Tyrosinkinase (MuSK). ARGX-119 fördert die Dimerisierung und Phosphorylierung von MuSK, was die Signalübertragung an der neuromuskulären Synapse verbessert und die Muskelfunktion wiederherstellt.26,27
Einen weiteren Ansatz, mit dem Ziel, die Erregbarkeit der Muskelzellen zu erhöhen, untersucht die Phase-II-Studie SYNAPSE-SMA zum CIC-1-Chloridkanal-Inhibitor NMD670.28 Im präklinischen Setting konnte NMD670 bereits die Reaktionsfähigkeit der Muskeln in Bezug auf schwache Signale verstärken, die neuromuskuläre Übertragung verbessern und die Funktion der Skelettmuskulatur wiederherstellen.29
Myotone Dystrophie Typ 1
Die myotone Dystrophie Typ 1 beim Kind
Die myotone Dystrophie Typ 1 (DM1) ist eine erbliche Multisystemerkrankung, die unter anderem die Muskeln, das Gehirn, das endokrine System und den Gastrointestinaltrakt betrifft. Ursächlich ist die Expansion einer instabilen CTG-Triplet-Repeat-Sequenz im Dystrophia-myotonica-Protein-Kinase(DMPK)-Gen, wobei die Länge der CTG-Repeats mit dem Schweregrad assoziiert ist und von Generation zu Generation zunimmt.30 Durch die Repeat-Verlängerungen werden unter anderem die Aktivität von Transkriptionsfaktoren und das Spleißen der RNA beeinflusst, womit die Expression verschiedener Gene verändert wird.31 Die DM1 kann in eine kongenitale, kindliche, adulte und Late-Onset-Form unterteilt werden, wobei ein früher Krankheitsbeginn zu größeren Einschränkungen führt.32,33 Bei der kongenitalen Form kommt es zu einer Hypotonie und Schwäche, einer respiratorischen Insuffizienz und Schluckstörungen. Auch frühe gastrointestinale und orthopädische Probleme zählen zu den Symptomen. Bei älteren Kindern treten neben Muskelschwäche und Aktionsmyotonie oftmals eine Facies myopathica oder eine Ptosis auf.33
Kognition und nichtmuskuläre Symptome in der DM1-Behandlung
„Die DM1 betrifft aber vor allem auch die Kognition und führt zu einer Minderung der Intelligenz und Aufmerksamkeitsdefiziten“, erklärt Priv.-Doz. Dr. Matthias Baumann von der Medizinischen Universität Innsbruck. Daher sollte die Behandlung eine detaillierte entwicklungsneurologische und neuropsychologische Testung und eine frühzeitige motorische und mentale Förderung beinhalten. Diese kann oftmals auch eine psychologische und kinder- und jugendpsychiatrische Betreuung umfassen.34 Auch die vorliegenden Schlafstörungen haben großen Einfluss auf das Alltagsbefinden und sollten von einem Spezialisten betreut werden.35 Oft bleiben gastrointestinale Symptome unerkannt und es kommt häufig zu urologischen Störungen, die unter anderem ein Toilettentraining bei Kleinkindern erschweren.36
Die myotone Dystrophie Typ 1 beim Erwachsenen
Die DM1 ist die häufigste Muskelerkrankung bei Erwachsenen, mit einer Prävalenz von 1:8000. Erwachsene weisen aber im Vergleich zu den DM1-Frühformen eine weniger schwere Symptomatik auf.37 Oftmals haben Betroffene gar keine Symptome und die Erkrankung fällt erst in Folgegenerationen auf. Symptome wie eine Fußheber- oder Fingerflexorschwäche, Katarakt, Ptose, Dysarthrie und erhöhte Gamma-GT-Werte können auf die Erkrankung hinweisen. Aufgrund der Erblichkeit von DM1 sollte im Verdachtsfall auch ohne ein Vorliegen von Beschwerden eine genetische Testung erfolgen und ein EMG angefertigt werden, das DM1-typische myotone Entladungen zuverlässig detektieren kann.38
Die Sterblichkeit von erwachsenen DM1-Patient:innen ist erhöht, wobei die Todesursache mehrheitlich auf respiratorische und kardiale Komplikationen zurückzuführen ist.39 „DM1 ist auch im Erwachsenenalter eine Multiorganerkrankung und gerade die respiratorischen und kardialen Dysfunktionen können ohne Beteiligung anderer Muskelgruppen auftreten“, betont Dr. Corinne Horlings von der Medizinischen Universität Innsbruck. Hier empfehlen sich eine regelmäßige Verlaufskontrolle und eine frühzeitige Behandlung.40,41
Krankheitsmodifizierende Therapieansätze bei DM1
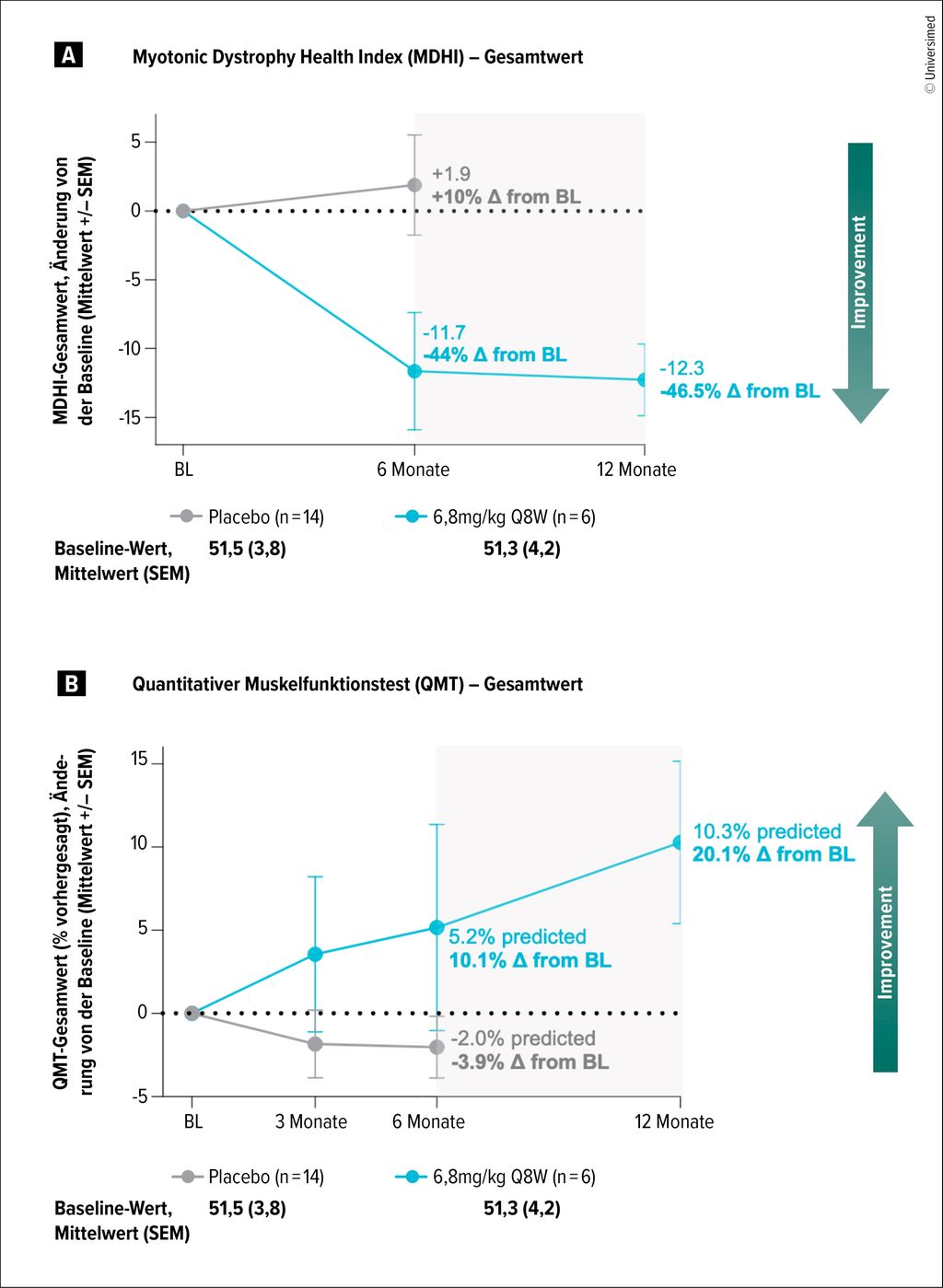
Abb. 2: Funktionale Verbesserungen bei Patient:innen mit myotoner Dystrophie Typ 1 unter DYNE-101 (modifiziert nach Sansone V et al. 2025)42
Auch bei DM1 rücken krankheitsmodifizierende Therapieansätze in den Fokus. In der Phase-I/II-Studie ACHIEVE wurde der Nutzen von DYNE-101 untersucht, einem Wirkstoff, der aus einem Transferrinrezeptor-1-bindenden Antikörper-Fragment besteht, das mit einem Antisense-Oligonukleotid (ASO) konjugiert ist. Durch die DM1-typischen CTG-Repeat-Verlängerungen entstehen DMPK-RNA-Aggregate, die unter anderem Spleißfaktoren wie MBNL1 dauerhaft binden und somit die Spleißmaschinerie blockieren und die Genexpression verändern.31 DYNE-101 bindet die DMPK-RNA-Aggregate und rekrutiert die RNase H1, welche die mRNA-Aggregate spaltet und die gebundenen Proteine freisetzt. So konnten unter DYNE-101 bereits weitreichende funktionale und kognitive Verbesserungen erzielt werden, auf deren Basis kürzlich die Phase-III-Studie HARMONIA angekündigt wurde (Abb. 2).42–44
Quelle:
Vorträge von Dr. Anna Wiesenhofer, Dr. Astrid Eisenkölbl, Priv.-Doz. Dr. Matthias Baumann und Dr. Corinne Horlings auf dem UpDate Muskelforschung vom 27.–28.2.2026 in Wien
Literatur:
1 Pichavant C et al.: Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther 2011; 19(5): 830-40 2 Bladen CL et al.: The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 2015; 36(4): 395-402 3 Kalman L et al.: Quality assurance for Duchenne and Becker muscular dystrophy genetic testing: development of a genomic DNA reference material panel. J Mol Diagn 2011; 13(2): 167-74 4 Emery AE.: The muscular dystrophies. Lancet 2002; 359(9307): 687-95 5 Bushby K et al.: Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010; 9(1): 77-93 6 Duan D et al.: Duchenne muscular dystrophy.Nat Rev Dis Primers 2021; 7(1): 13 7 Griggs RC et al.: Efficacy and safety of deflazacort vs prednisone and placebo for Duchenne muscular dystrophy. Neurology 2016; 87(20): 2123-31 8 Mah JK et al.: Efficacy and safety of Vamorolone in Duchenne muscular dystrophy: a 30-month nonrandomized controlled open-label extension trial. JAMA Netw Open 2022; 5(1): e2144178 9 Dang UJ et al.: Efficacy and safety of Vamorolone over 48 weeks in boys with duchenne muscular dystrophy: a randomized controlled trial. Neurology 2024; 102(5): e208112 10 Pressemitteilung Santhera vom 15.01.26. Abrufbar unter: https://www.santhera.de/assets/files/press-releases/260115-Santhera-Swissmedic-approval-FINAL-Ger.pdf 11 Fachinformation Duvyzat, Stand 10/2025 12 Aartsma-Rus A: Histone deacetylase inhibition with givinostat: a multi-targeted mode of action with the potential to halt the pathological cascade of Duchenne muscular dystrophy. Front Cell Dev Biol 2025; 12: 1514898 13 Mercuri E et al.: Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 2024; 23(4): 393-403 14 Mendell JR et al.: AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial. Nat Med 2025; 31(1): 332-41 15 Etxaniz U et al.: AOC 1044 induces exon 44 skipping and restores dystrophin protein in preclinical models of Duchenne muscular dystrophy. Nucleic Acids Res 2025; 53(6): gkaf241 16 Chong LC et al.: Drug discovery of spinal muscular atrophy (SMA) from the computational perspective: a comprehensive review. Int J Mol Sci 2021; 22(16): 8962 17 Parsons JA et al.: Remaining burden of spinal muscular atrophy among treated patients: a survey of patients and caregivers. Ann Clin Transl Neurol 2025; 12(10): 2020-35 18 de Lemus M et al.: Identification of the most relevant aspects of spinal muscular atrophy (SMA) with impact on the quality of life of SMA patients and their caregivers: the PROfuture project, a qualitative study. J Patient Rep Outcomes 2024; 8(1): 78 19 Finkel RS et al.: Risdiplam for prenatal therapy of spinal muscular atrophy.N Engl J Med 2025; 392(11): 1138-40 20 Finkel RS et al.: In utero risdiplam for severe SMA: a case series. WMS 2025; Abstract 04LBO 21 Presseinformation Biogen vom 12.1.26, abrufbar unter: https://investors.biogen.com/news-releases/news-release-details/biogen-receives-european-commission-approval-high-dose-regimen 22 Finkel RS et al.: High-dose nusinersen for spinal muscular atrophy: a phase 3 randomized trial. Nat Med 2026; 32(3): 1095-1104 23 Finkel RS et al.: DEVOTE study exploring higher dose of nusinersen in spinal muscular atrophy: study design and part A results. J Neuromuscul Dis 2023; 10(5): 813-23 24 Crawford T et al.: Exploring higher doses of nusinersen in spinal muscular atrophy: final results from parts B and C of the 3-Part DEVOTE Study. WMS 2024; Abstract 710LBP 25Finkel RS et al.: DEVOTE part C results: exploring higher doses of Nusinersen in Nusinersen-experienced participants with spinal muscular atrophy. CURE SMA 2025; Abstract 28 26 Vanhauwaert R et al.: ARGX-119 is an agonist antibody for human MuSK that reverses disease relapse in a mouse model of congenital myasthenic syndrome. Sci Transl Med 2024; 16(765): eado7189 27 Coppieters S (Argenx): A phase 2 double-blinded, randomized, placebo-controlled study to assess the safety, tolerability, efficacy, pharmacokinetics, and immunogenicity of intravenous administration of ARGX-119 in pediatric participants aged 5 to less than 18 years with spinal muscular atrophy. ClinicalTrials.gov ID: NCT07287982 28 NMD Pharma: A phase 2, randomised, double-blind, placebo-controlled, 2-way crossover study to evaluate the efficacy, safety, and tolerability of NMD670 in ambulatory adults with type 3 spinal muscular atrophy. ClinicalTrials.gov ID: NCT05794139 29 Skov M et al.: The ClC-1 chloride channel inhibitor NMD670 improves skeletal muscle function in rat models and patients with myasthenia gravis. Sci Transl Med 2024; 16(739): eadk9109 30 Rahm L et al.: Myotonic dystrophy type 1: clinical diversity, molecular insights and therapeutic perspectives. Nat Rev Neurol 2025; 21(11): 623-41 31 Llamusí B et al.: Molecular effects of the CTG repeats in mutant dystrophia myotonica protein kinase gene. Curr Genomics 2008; 9(8): 509-16 32 Kuntawala DH et al.: Multisystem symptoms in myotonic dystrophy type 1: a management and therapeutic perspective. Int J Mol Sci 2025; 26(11): 5350 33 Lagrue E et al.: A large multicenter study of pediatric myotonic dystrophy type 1 for evidence-based management. Neurology 2019; 92(8): e852-e865 34 Sweere DJJ et al.: Cognitive phenotype of childhood myotonic dystrophy type 1: a multicenter pooled analysis. Muscle Nerve 2023; 68(1): 57-64 35 Quera Salva MA et al.: Sleep disorders in childhood-onset myotonic dystrophy type 1. Neuromuscul Disord 2006; 16(9-10): 564-70 36 Maagdenberg SJM et al.: Impact of gastrointestinal and urological symptoms in children with myotonic dystrophy type 1. Neuromuscul Disord 2024; 35: 1-7 37 De Antonio M et al.: Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 2016; 172(10): 572-80 38 Drost G et al.: Myotonic discharges discriminate chloride from sodium muscle channelopathies. Neuromuscul Disord 2015; 25(1): 73-80 39 Gagnon C et al.: Neuromuscular Disord 2025; 53(Suppl. 105941): Abstract 375P 40 Kuntawala DH et al.: Multisystem symptoms in myotonic dystrophy type 1: a management and therapeutic perspective. Int J Mol Sci 2025; 26(11): 5350 41 Schnelder-Gold C et al.: Myotone Dystrophien, nicht dystrophe Myotonien und periodische Paralysen, S1-Leitlinie, 2023. In: Deutsche Gesellschaft für Neurologie, Hrsg. Leitlinien für Diagnostik und Therapie in der Neurologie. Online: www.dgn.org/leitlinien (abgerufen am 24.3.2026) 42 Sansone V et al.: WMS 2025; Poster 380 43 Dyne Clinical Trials: a randomized, placebo-controlled, multiple ascending dose study assessing safety, tolerability, pharmacodynamics, efficacy, and pharmacokinetics of DYNE-101 administered to participants with Myotonic Dystrophy Type 1. ClinicalTrials.gov ID: NCT05481879 44 https://www.dyne-tx.com/clinical-trials/
Das könnte Sie auch interessieren:

Ist die ketogene Diät eine Präzisionsmedizin?
Die ketogenen Ernährungstherapien sind etablierte Behandlungsformen bei Epilepsie. Während sie primär bei therapierefraktären pädiatrischen Epilepsien eingesetzt werden, finden sie ...

Neues aus der Alzheimer’s Disease Drug Development Pipeline
Mit der weltweiten Zulassung der Amyloidantikörper Lecanemab und Donanemab ist erstmals eine kausale Behandlung der Alzheimerkrankheit möglich geworden. Die Behandlung setzt an der ...
Neuroonkologie: neue therapeutische Möglichkeiten
Lange musste in der Neuroonkologie auf eine Medikamenten-zulassung gewartet werden. Dank intensiver Forschung mit detaillierter molekularer Tumorcharakterisierung konnte im letzten Jahr ...