
Hämatologische Labordiagnostik in der Arztpraxis
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Laborresultate liefern wichtige Zusatzinformationen, die Interpretation der Werte gestaltet sich jedoch nicht immer einfach. Schritt für Schritt und mithilfe von Kasuistiken zeigte Prof. Dr. med. Lorenzo Alberio, Klinik für Hämatologie und hämatologisches Zentrallabor CHUV, in einem Workshop am Frühjahrskongress der SGAIM das Vorgehen bei der Labordiagnostik auf.
Keypoints
-
Bei Patienten mit unklarer,persistierender Thrombozytose muss eine essenzielle Thrombozythämie erwogen und abgeklärt werden.
-
Die echte Thrombozytopenie kombiniert mit einer Anämie plus Hämolysezeichen und Fragmentozyten im Blutausstrich ist ein Notfall.
-
Zeigt das Labor eine Panzytopenie, muss immer ein Gerinnungsstatus verlangt und eine disseminierte intravasale Gerinnung ausgeschlossen werden.
-
Niedermolekulare Heparine und Faktor-Xa-Inhibitoren können in hoher Konzentration vorhanden sein, ohne die Gerinnungsanalytik in den pathologischen Bereich zu verschieben.
Abklärung einer Thrombozytose
Eine Thrombozytose (>450000/µl) kann eine Vielzahl von Ursachen haben. Mithilfe eines Blutausstrichs muss zunächst das Vorliegen einer Pseudothrombozytose ausgeschlossen werden. Diese kann durch Mikroerythrozyten, Tumorzellfragmente oder Kryoglobuline verursacht werden, die fälschlicherweise als Thrombozyten gezählt werden.1Als Differenzialdiagnosen bei einer echten Thrombozytose kommen neben den seltenen hereditären und primär erworbenen Formen wie beispielsweise den myeloproliferativen Neoplasien sekundäre Ursachen wie Eisenmangel, Infekte, Entzündungen oder Asplenie infrage. Die laborchemische Abklärung in der internistischen Hausarztpraxis umfasst Blutbild, Entzündungsparameter und Eisenstatus. Sorgen diese nicht für Klarheit, sollte der Patient einem Hämatologen oder einer Hämatologin für eine ausgedehnte Diagnostik zugewiesen werden. «Das ist wichtig, weil im Kontext einer langjährigen Thrombozytose nicht selten thromboembolische Komplikationen auftreten», sagte Prof. Dr. med. Lorenzo Alberio.
In einem kürzlich im European Journal of Internal Medicine publizierten «Letter to the Editor» wies knapp die Hälfte der beschriebenen Patienten mit einer persistierenden Thrombozytose eine essenzielle Thrombozythämie mit initialen thrombotischen Manifestationen auf (n=20).2 Bei 15 Patienten war das thromboembolische Ereignis vor und bei 5 Patienten zum Zeitpunkt der Diagnose der essenziellen Thrombozythämie aufgetreten. Die Zeit vom Nachweis der Thrombozytose bis zum Auftreten eines ersten solchen Ereignisses betrug bei einer Thrombozytenzahl von >350000/µl im Median 21 Monate und bei einer Thrombozytenzahl >450000/µl im Median 8 Monate. Die Zeit vom Nachweis der Thrombozytose bis zur Diagnose einer essenziellen Thrombozythämie lag im Mittel bei 26 respektive 13 Monaten.
Abklärung einer Thrombozytopenie
Auch bei der Thrombozytopenie sollte zunächst an eine Pseudothrombozytopenie gedacht werden: «Diese tritt sehr viel häufiger auf als eine Pseudothrombozytose», sagte der Spezialist. Einen ersten Hinweis auf das Vorliegen einer Pseudothrombozytopenie liefert das sägezahnartige Histogramm im Ausdruck des Messgeräts (Abb. 1).3 Bestätigt wird der Verdacht durch den Nachweis von Thrombozytenaggregaten im Blutausstrich.4 Das Phänomen wird als EDTA-induzierte Pseudothrombozytopenie beschrieben und ist eine Folge der zu langen Lagerung zwischen Blutentnahme und Labormessung. Bei der Verwendung von Blutentnahmeröhrchen mit Natriumcitrat tritt das Phänomen in der Regel nicht auf.5
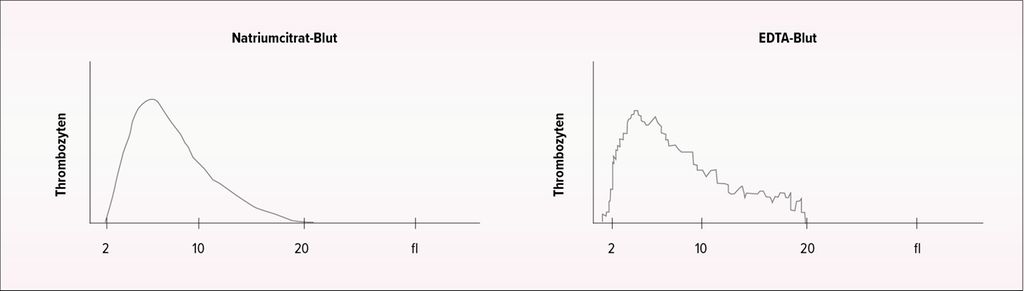
Abb. 1: Typisches Histogramm bei EDTA-induzierter Pseudothrombozytopenie (rechts). Im Vergleich dazu das Histogramm einer Natriumcitrat-Blutprobe bei dem gleichen Patienten (links) (adaptiert nach Nagler et al., 2014)3
Das Vorgehen zur Abklärung bei Verdacht auf eine echte Thrombozytopenie erklärte der Spezialist anhand der Krankengeschichte von Domenica, einer 41-jährigen Patientin, die wegen einer Bizytopenie und eines vergrösserten Pankreas hospitalisiert wird. Seit etwa einer Woche ist der Urin der Patientin dunkel verfärbt. Wenige Tage vor dem Spitaleintritt treten perimalleoläre Petechien auf, zusätzlich leidet die Patientin an Übelkeit, Bauch- und Kopfschmerzen. Das Blutbild zeigt eine Thrombozytopenie (13000/µl) und eine normozytäre normochrome Anämie. Die hyperregenerativen Retikulozyten bestätigen, dass es sich nicht um ein Produktionsproblem, sondern um einen Verlust von Erythrozyten handelt. Die stark erhöhte LDH deutet auf eine hämolytische Anämie hin. Ob diese mechanisch oder immunologisch ausgelöst wurde, soll ein direkter Antiglobulintest (Coombs-Test) klären. Das Ergebnis ist negativ, es handelt sich somit um eine mechanische Ursache. Die Vielzahl von Mikrosphärozyten – eine Subgruppe von Fragmentozyten – im Blut liefert den Hinweis auf eine thrombotische Mikroangiopathie entweder infolge eines hämolytisch-urämischen Syndroms oder einer thrombotisch-thrombozytopenischen Purpura (TTP).6 «Die Diagnose einer TTP ist ein Notfall, die Mortalität liegt bei 80% und kann durch eine umgehende Plasmapherese bis auf ca. 20% reduziert werden», so Alberio. Durch die Behandlung mit den monoklonalen Antikörpern Rituximab und allenfalls Caplacizumab könne das Outcome weiter verbessert werden.7
Wann ist ein Gerinnungsstatus indiziert?
Mit der Geschichte von Giorgio leitete der Spezialist zum Hämostaselabor über. Der 72-Jährige leidet seit einiger Zeit an zunehmender Müdigkeit. Wegen eines Gichtanfalls wird er seit ca. 4 Wochen mit Colchicin behandelt. Bekannt sind eine Hyperferritinämie, die mit 3 Phlebotomien pro Jahr behandelt wird, und eine arterielle Hypertonie. Das Labor zeigt eine Panzytomie, mit normochromer normozytärer Anämie, Leukopenie und Thrombozytopenie. Der nachträglich angeforderte Gerinnungsstatus ergibt folgende Werte: Quick 30%, aPTT 33sec, Thrombinzeit (TZ) 20sec, Fibrinogen 0,5g/l.
Auffällig ist vor allem der niedrige Fibrinogenwert. Die Bestimmung der D-Dimere soll Aufschluss darüber geben, ob die Fibrinogenproduktion zu niedrig oder der Verbrauch zu hoch ist. Das Labor zeigt eine D-Dimer-Anzahl von 50600ng/ml, was für eine schwere disseminierte intravasale Gerinnung (DIC) spricht. Blutungszeichen gibt es nicht. Die anschliessende Knochenmarksuntersuchung ergibt eine akute Promyelozytenleukämie (AML M3). «Bei einer Panzytopenie muss immer ein Gerinnungsstatus verlangt und eine DIC ausgeschlossen werden», sagte der Spezialist. Das gemeinsame Auftreten von Panzytopenie und Verbrauchskoagulopathie sollte zur Verdachtsdiagnose einer AML M3 führen. Aber aufgepasst: Das Auftreten von Panzytopenien wird auch im Zusammenhang mit Colchicin beschrieben. In diesem Fall eine «falsche Fährte», die zur Verschleppung der Diagnose hätte führen können. «Deshalb sollte man die Differenzialdiagnose immer möglichst breit stellen», lautete das Fazit des Referenten.
Abklärung bei neu aufgetretener Blutungsneigung
Eine kurze Rekapitulation der Gerinnung verdeutlichte den zahlreichen Zuhörern noch einmal die beiden Gerinnungswege, die sich (auch symbolisch) bei Faktor X kreuzen und mit 3% (extrinsisch) und 97% (intrinsisch) mit sehr unterschiedlichen Proportionen zur Thrombinbildung beitragen (Abb. 2).
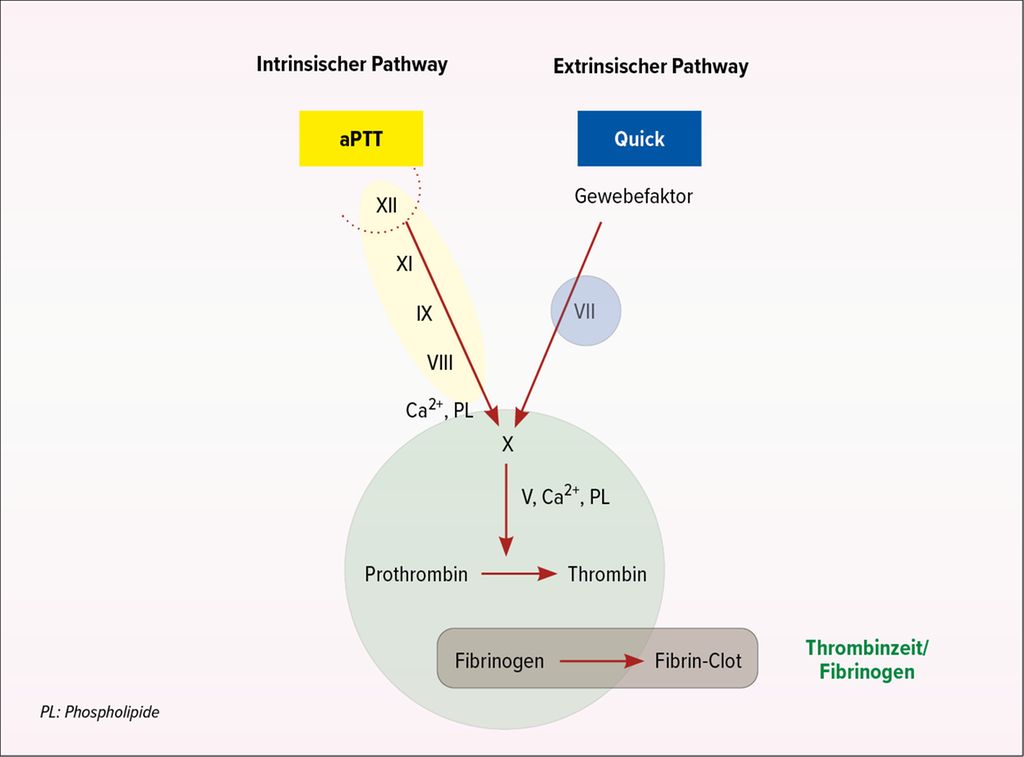
Abb. 2: Gerinnungswege und zugehörige Gerinnungstests (Quelle: Prof. Dr. med. L. Alberio)
Anhand der vier Gerinnungsparameter – aPTT, Quick, Thrombinzeit (TZ) und Fibrinogen – lässt sich die Ursache für eine Blutung identifizieren. Ist nur der Quick auffällig und sind die übrigen Parameter normal, ist der Faktor VII der Verursacher des Problems. Bei einer isoliert verlängerten aPTT kann es sich um einen Mangel der Faktoren XI, IX, VIII oder von Willebrand handeln. Der Faktor XII ist von der normalen Gerinnung ausgeschlossen. Eine isoliert verlängerte aPTT kann zudem die Folge einer erworbenen Hämophilie aufgrund von Autoantikörpern gegen Faktor VIII oder Lupus-Antikoagulans sein. Letzteres führt zu einem vermehrten Auftreten von Thrombosen. Für die Unterscheidung muss kein Speziallabor hinzugezogen werden, sie gelingt mit einfachen Mitteln in der internistischen Allgemeinarztpraxis. Man mischt dazu das Plasma des Patienten mit normalem Blutplasma (Mischverhältnis 1:1, Beurteilung sofort und nach 1 Stunde bei 37°C). Führt die Zugabe des normalen Plasmas zu einer Korrektur der aPTT, handelt es sich um einen Mangel. «Wenn sich dagegen die aPTT im Verlauf der Inkubation verlängert, dann haben Sie den Faktor-VIII-Inhibitor entdeckt», sagte der Spezialist.
Dazu eine weitere Kasuistik: die Geschichte von Falco, einem Patienten mit erworbener Blutungsneigung und einer isolierten prolongierten aPTT, die mehrfach im Abstand von mehreren Wochen bestätigt wurde. Eine Behandlung des 66-Jährigen mit FFP und anschliessende Kontrolle der aPTT wurde in Betracht gezogen, dazu kam es aber nicht. Einige Zeit später wurde er notfallmässig wegen einer Hirnblutung hospitalisiert, an deren Folgen er schliesslich starb.
«Neu erworbene Blutungsneigungen werden häufig erst mit Verzögerung diagnostiziert», sagte der Spezialist. Um dramatische Verläufe wie diesen zu verhindern, sollte man bei Patienten mit neu aufgetretener Blutungsneigung und isoliert verlängerter aPTT an eine erworbene Hämophilie denken und einen Mischversuch machen oder das Labor darum bitten. Ist das nicht möglich, sollte der Patient auf den Notfall überwiesen werden.
Häufige Gerinnungsstörungen
Die Verlängerung von Quick und aPTT bei gleichzeitig normaler Thrombinzeit (TZ) deutet auf eine Synthesestörung oder einen Verbrauch hin.
Hier liefert die Bestimmung der Gerinnungsfaktoren VII, X, V und Prothrombin (Faktor II) weitere Informationen (Abb. 3). Ein isoliert erhaltener FaktorV deutet auf ein Vitamin-K-Defizit hin. Sind alle vier Faktoren (VII, X, V, II) zu niedrig, könnte eine Hepatopathie die Ursache sein. Das typische Labor bei einer DIC zeigt einen ausgeprägten Mangel der Faktoren VII und V, während die Faktoren X und II entweder normal oder leicht erniedrigt sind.
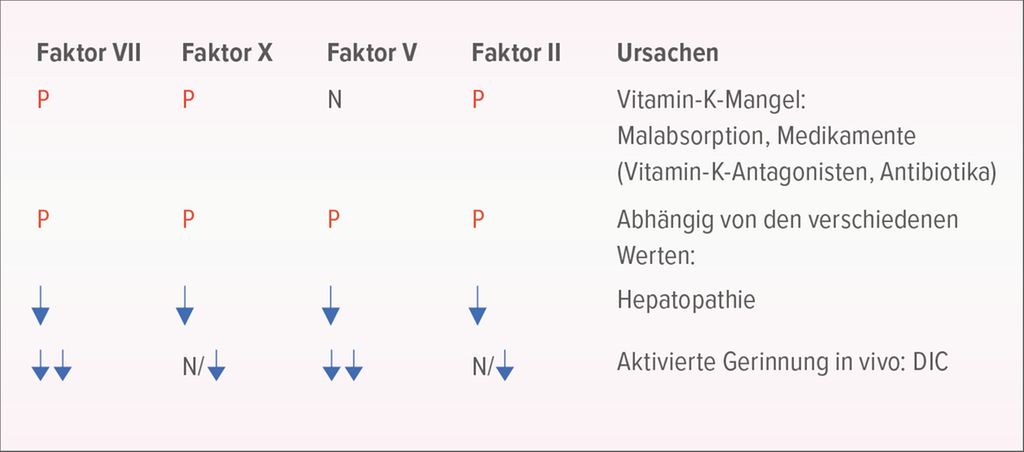
Abb. 3: Labor-Work-up bei niedrigem Quick plus verlängerter aPTT und normaler Thrombinzeit (Quelle: Prof. Dr. med. L. Alberio)
Ist neben Quick und aPTT auch die TZ verlängert, sollte zuerst an die Antikoagulation mit einem direkten Thrombininhibitor (FaktorIIa) gedacht werden – im ambulanten Setting vermutlich mit Dabigatran.
Wenn die Gerinnung normal ist und der Patient blutet
Blutet der Patient trotz einer normalen Gerinnungsanalytik, kommen als Ursache eine gestörte Thrombozytenfunktion oder eine von-Willebrand-Erkrankung, in seltenen Fällen Kollagenopathien, Faktor-XIII-Mangel, Hyperfibrinolyse, die Behandlung mit niedermolekularen Heparinen oder Faktor-Xa-Inhibitoren (Apixaban, Rivaroxaban, Edoxaban) infrage. Letztere können in hoher Konzentration vorhanden sein, ohne die Gerinnungswerte in den pathologischen Bereich zu verschieben.
Quelle:
Frühjahrskongress der SGAIM, 10.–12. Mai, Basel
Literatur:
1 Alberio L: Do we need antiplatelet therapy in thrombocytosis? Pro. Hamostaseologie 2016; 36: 227-40 2 Stalder G et al.: Long-standing thrombocytosis often precedes thromboembolic complications heralding the diagnosis of essential thrombocythemia. Eur J Intern Med 2023; 107: 110-2 3 Nagler M et al.: A case of EDTA-dependent pseudothrombocytopenia: simple recognition of an underdiagnosed and misleading phenomenon. BMC Clin Pathol 2014; 14: 19 4 Amoruso M et al.: EDTA-related degranulation mimicking Storage Pool Disease. Am J Hematol 2018; 93: 1192-93 5 Greinacher A, Selleng S: How I evaluate and treat thrombocytopenia in the intensive care unit patient. Blood 2016; 128: 3032-42 6 Lesesve J-F et al.: Fragmented red blood cells automated measurement is a useful parameter to exclude schistocytes on the blood film. Int J Lab Hematol 2012; 34: 566-76 7 Mele C et al.: Hemolytic uremic syndrome. Semin Immunpathol 2014; 36: 399-420
Das könnte Sie auch interessieren:

Planetary Health als hausärztliche Aufgabe: Gesundheit im Zeitalter der ökologischen Krise
Hausärzt:innen sind geradezu prädestiniert, um den Gedanken der Planetary Health umzusetzen, denn sie verfügen über das Vertrauen, die Reichweite und die Handlungsspielräume, um ...
Mehr als nur eine trockene Angelegenheit: Die Sjögren-Erkrankung im Fokus
Die Sjögren-Erkrankung (SjD) ist eine chronisch-entzündliche, systemische Autoimmunerkrankung mit hoher klinischer Variabilität. Sie gehört zur Familie der Kollagenosen, zu der ...

Tinnitusmanagement 2025
Die Behandlung von Tinnitus hat sich in den letzten Jahren weiterentwickelt. Die AWMF-Leitlinien zum Tinnitusmanagement bieten evidenzbasierte Empfehlungen zur Diagnostik und Therapie ...