
Herausforderung in Diagnose und Therapie
Während Phäochromozytome aus dem Nebennierenmark stammen, sind adrenokortikale Karzinome (ACC) Tumoren der Nebennierenrinde. Paragangliome können in den Paraganglien zwischen Kopf- und Beckenbereich auftreten. In seinem Vortrag im Rahmen desCUO behandelte Univ.-Pof. Priv.-Doz. Dr. Mesut Remzi, Medizinische Universität Wien, die Herausforderungen in der Diagnose, Charakteristika in der Bildgebung und die Therapie dieser beiden Entitäten.
Keypoints
-
Zum Nachweis Katecholamin-produzierender PPGL eignet sich der 24h-Urin oder Blutplasma.
-
Um ein Phäochromozytom ausschließen zu können, ist ein PET-CT bei unklarer heller Läsion im CT unabdingbar.
-
18F-FDG oder 18F-DOPA sind dem 123I-MIBG in der Identifizierung von Paragangliomen überlegen.
-
Ein genetisches Screening ist aufgrund der Häufigkeit von genetischen Mutationen bei PPGL anzuraten.
-
Zur Diagnose des ACC ist es wichtig, den Hormonspiegel zu überprüfen.
-
Zum ACC-Staging eignet sich am besten 18F-FDG-PET.
Phäochromozytome und Paragangliome (PPGL)
Paragangliome (PGL) und Phäochromozytome (Phäo) zählen zu ein- und derselben Krankheitsentität und werden unter der Abkürzung PPGL zusammengefasst. Sie sind potenziell maligne, und zwar dann, wenn im Zuge des Follow-ups (FU) Metastasen detektiert werden. Das Komplizierte daran ist, dass es keine klinischen, laborspezifischen oder histologischen Charakteristika gibt, die eine Einstufung als maligne ermöglichen würden. Darüber hinaus zeichnen sie sich durch sehr unspezifische Symptome aus, die durch Stress, mechanischen Druck, Medikamente oder bestimmte Nahrungsmittel getriggert werden können.1 Zu den klassischen Symptomen – sie treten nur in 19% der Fälle auf – zählen Tachykardien, Kopfschmerzen und Schwitzen.2
Risikofaktoren für Malignität sind eine Tumorgröße >5cm, ein Ki-67-Index >3%, ein um das 3-Fache erhöhter Metoxytyraminspiegel, ein PASS(„pheochromocytoma of the adrenal gland scaled score“)-Score >6 sowie ein GAPP(„pheochromocytoma of the adrenal gland scaled score“)-Score >7.3 Zusätzlich muss auf das Vorliegen einer SDHB-Mutation („succinate dehydrogenase B mutation“) getestet werden.4
Bestimmung der Katecholaminproduktion
PPGL produzieren die Katecholamine Noradrenalin (NA), Adrenalin (A) oder Dopamin (D), die zu ihren Metaboliten Normetanephrin (NA), Metanephrin (M)und Methoxytyramin (3MT) umgewandelt werden. Letztere sind einfacher zu detektieren. Außerdem sollte der Level von Chromogranin A bestimmt werden, da es im Fall einer Erhöhung als Tumormarker gewertet werden kann. Die Hälfte der Phäo produzieren NA + A, der Rest nur NA. Abdominale PGL produzieren nur NA und wenig D. Es gibt auch thorakale und PGL im Kopf, die meistens keine Katecholamine produzieren. Nachgewiesen werden können die Katecholaminmetaboliten im 24h-Urin oder besser noch – da mit einer höheren Sensitivität (97%) und Spezifität (91%) – im Plasma.
Bildgebung und Histologie
Sollte eine Läsion >20 HU (Hounsfield- Einheiten) im CT ohne Kontrastmittel aufweisen, besteht der Verdacht auf das Vorliegen eines Phäo oder eines ACC (adrenokortikales Karzinom). Meistens wird zusätzlich eine Vergrößerung der Blutgefäße beobachtet. Gemäß Leitlinien wird ein CT-Scan auf das Vorliegen eines ACC mit einem 10-minütigen Kontrastmittel-Wash-out empfohlen, wobei die Sensitivität dabei bei 88–100% liegt, die Spezifität bei 70–80%. Ein CT-Scan auf das Vorliegen eines PGL oder von Metastasen ist weniger sensitiv.3 Ebenso kann ein MRT mit T2-Gewichtung durchgeführt werden. Ein Phäo kann sich hier zystisch und hämorrhagisch bzw. entweder homogen oder heterogen präsentieren. Bei Feststellung von hellen Läsionen handelt es sich am wahrscheinlichsten um ein Phäo, sollte dies aber nicht der Fall sein, kann eine Phäo nicht automatisch ausgeschlossen werden. „Aus diesem Grund ist die funktionelle Bildgebung mittels PET(Positronenemissionstomografie)-CT in diesem Setting so wichtig“, betonte Prof. Remzi. Dabei müssen aber verschiedene Faktoren berücksichtigt werden. Nicht bei jedem Patienten ist folglich eine PET-CT-Untersuchung indiziert. Ein PET-CT mit 18F-FDG (Fluordesoxyglucose) oder 18F-DOPA ist besser für die Identifizierung von PGL, Metastasen und von SDH-Mutation-assoziierten Tumoren geeignet als eine Szintigrafie mit 123I-MIBG (I-Meta-Iodobenzylguanidin). Für die Diagnose von sporadischem und kleinem Phäo wiederum ist ein DOPA-PET am besten – sogar besser als ein DOTA-PET – geeignet. Ein DOTA-PET ist die optimale Methode für die Diagnose von SDH-mutierten Tumoren und Keimbahnmutationen. Dabei wird ein an DOTAgekoppeltes Peptid wie DOTATET, DOTANOC oder DOTATOC als Chelator verwendet.6
PPGL sind neuroendokrine Tumoren, wobei 5–10% der solitären Phäo hereditär sind. Bei allen anderen Formen des Phäo werden hereditäre Mutationen in 40–70% der Fälle detektiert. Zu den häufigsten Mutationen zählen SDHB (10%), SDHD (9%), VHL (7%), RET (6%) und NF1 (3%).7 „Man sollte sich demnach auf das genetische Screening fokussieren, was auch durch umfassende Studien bestätigt ist“, unterstrich Remzi. Lenders JWM et al. liefern hierzu den diagnostischen Algorithmus, um bei Diagnose eines PPGL mit oder ohne Mutation die Wahl der funktionellen Bildgebung zu treffen (Abb. 1).8 Histologisch ist eine eindeutige Differenzierung zwischen maligne und benigne nicht möglich, jedoch können der PASS- und der GAPP-Score zur Prädiktion der Malignität und als Anhalt im FU genutzt werden.
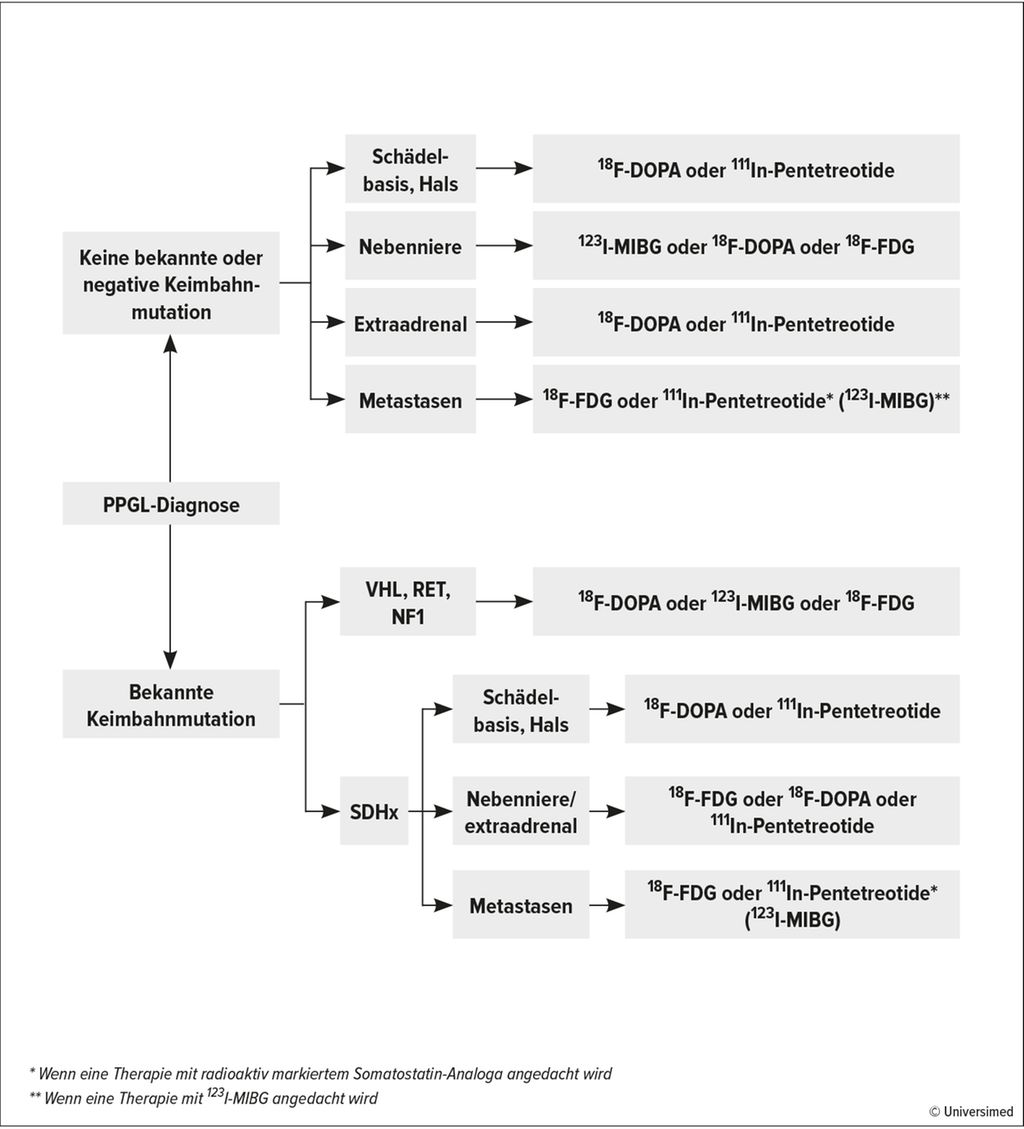
Abb. 1: Algorithmus zur Entscheidung für eine funktionelle Bildgebung bei Patienten mit nachgewiesenem PPGL (nach Lenders JWM et al. 2014)8
Operation als Therapie der Wahl
Das präoperative Management stellt insofern eine Herausforderung dar, als die Patienten an Hypertonie und niedrigem Blutvolumen leiden. Die Gabe von α-Blockern wird zwar in sämtlichen Leitlinien empfohlen, jedoch liegt nur sehr geringe Evidenz dafür vor:11 Es wurde nur eine vergleichende Studie dazu durchgeführt und in dieser konnte kein Unterschied hinsichtlich der Effekte nachgewiesen werden.12 „Wir diskutieren bei jedem Patienten individuell, ob wir α-Blocker verabreichen. Fallweise setzen wir auch Kalziumantagonisten ein“, berichtete Remzi. Auch in der Entscheidung zwischen einer kompletten (einseitigen) vs. eine subtotale Adrenalektomie muss differenziert vorgegangen werden: „Insbesondere, wenn ein Patient eine genetische Alteration aufweist, sollte die partielle Nebennierenresektion angestrebt werden, da ein hohes Risiko besteht, auf der kontralateralen Seite auch ein PPGL zu entwickeln und dann überhaupt kein gesundes Nebennierenparenchym mehr vorliegen würde“, erklärte Remzi.
Adrenokortikales Karzinom (ACC)
ACC zählen wie PPGL zu den Orphan Diseases. Ihr Management sollte demnach auf spezialisierte Zentren beschränkt sein, in denen ein multidisziplinäres Team an Spezialisten zur Verfügung steht.13 Bereits bei Diagnose weisen 40% der Patienten ein M1-Stadium auf und eine enorme Tumorgröße (im Median: >11cm). Hinsichtlich des Alters gibt es zwei Gipfel: <10 Jahren und in der 5.–6. Lebensdekade, wobei Erwachsene viel aggressivere Tumoren entwickeln als Kinder. Im Fall von ACC löst die Tumorgröße die Symptome aus. 40% der ACC sind hormonaktiv, 60% autokrin aktiv. „Wichtig ist es, immer die Cortisolspiegel und allgemein die in diesem Zusammenhang relevanten Hormonspiegel zu überprüfen, denn 50% der Patienten haben ein Cushingsyndrom und nach Resektion des Tumors könnte eine Addisonkrise auftreten“, betonte Remzi.
Bildgebung
Für die Diagnose sollte zur Evaluierung des Vorliegens von Lebermetastasen eine CT- oder MRT-Untersuchung durchgeführt werden. Für das Staging ist ein 18F-FDG-PET die optimale Methode. Sie geht mit einer Sensitivität von 97% und einer Spezifität von 91% einher. Wenn die FDG-Aufnahme niedriger als in der Leber ist, handelt es sich um einen benignen Tumor, wenn sie höher ist, besteht der V. a. ein ACC. ACC zeigen sich in einer unregelmäßigen Form sowie einer inhomogenen Dichte. In einem nicht verstärkten CT weisen sie einen Schwächungskoeffizienten von 20 HU auf.
Komplette Resektion als Ziel
Eine inkomplette Resektion führt innerhalb eines Jahres zum Tod. In Abhängigkeit vom Stadium (meist liegt ein Stadium II vor) verschlechtert sich die Prognose: Gemäß den Ergebnissen einer niederländischen Studie betrug das mediane Überleben bei Stadium I/II 159 Monate, bei Stadium III 26 Monate und bei Stadium IV nur 5 Monate (Abb. 2).14 „Ein negativer Resektionsrand und das Stadium sind die stärksten Faktoren für das Überleben. Die komplette chirurgische Resektion ist das oberste Ziel!“, fasste Remzi zusammen. Jedoch ist auch bei einer R0-Resektion in 50–80% der Fälle mit einem Rezidiv zu rechnen. Aus diesem Grund ist die adjuvante Therapie so relevant. Zurzeit ist nur das oral verfügbare Zytostatikum Mitotane in der Indikation ACC zugelassen.15 Mitotane wurde bereits 2004 von der EMA (European Medicines Agency) zugelassen.16 In einer retrospektiven Studie zum Vergleich mit Observation konnte die signifikante Überlegenheit der Substanz hinsichtlich des rezidivfreien Überlebens bestätigt werden.17 Die Entscheidung, ob Mitotane jedoch als Monotherapie oder zusätzlich zu einer Chemotherapie und/oder einer Radiotherapie verabreicht wird, entscheidet sich je nach dem KI-67-Status.18
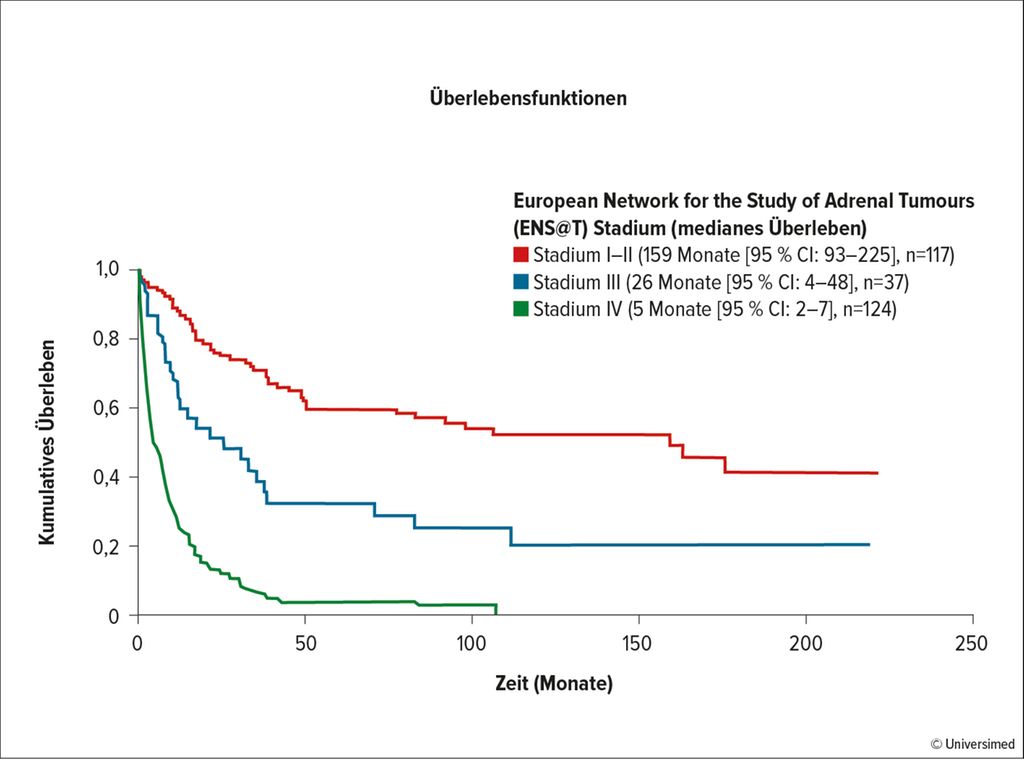
Abb. 2: AAC: Überlebensdauer in Abhängigkeit vom Stadium (nach Kerkhofs TMA et al. 2013)14
Quelle:
„Essentials of PPGL and ACC“, Vortrag von Priv.-Doz. Dr. Mesut Remzi im Rahmen des CUO (Controversies in Urologic Oncology) Scientific Meeting am 14. und 15. April 2023 in Wien
Literatur:
1 Lenders JWM et al.: Phaeochromocytoma. Lancet 2005; 366(9486): 665-75 2 Geroula A et al.: Pheochromocytoma and paraganglioma: Clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol 2019; 181(4): 409-20 3 Parenti G et al.: Updated and new perspectives on diagnosis, prognosis and therapy of malignant pheochromocytoma/paraganglioma. J Clin Oncol 2012; 872713 4 Gill AJ.: Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018; 72(1): 106-16 5 Fassnacht M et al.: Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020; 31(11): 1476-90 6 Han S et al.: Performance of 68Ga-DOTA-conjugated Somatostatin receptor-targeting peptide PET in detection of pheochromocytoma and paraganglioma: A systematic review and metaanalysis. J Nucl Med 2019; 60(3): 369-76 7 Neumann HPH et al.: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346(19): 1459-66 8 Lenders JWM et al.: Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J Clin Endocinol 2014; 99(6): 1915-42 9 Thompson LDR: Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002; 26(5): 551-66 10 Strong VE et al.: Prognostic indicators of malignancy in adrenal pheochromocytomas: Clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 2008; 143(6): 759-68 11 Neumann H et al.: Pheochromocytoma and paraganglioma. N Engl J Med 2019; 381(6): 552-65 12 Groeben H et al.: Perioperative α-receptor blockade in phaeochromocytoma surgery: An observational case series. Br J Anaesth 2017; 118(2): 182-9 13 Gajoux S, Mihai R: European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. Br J Surg 2017; 104(4): 358-76 14 Kerkhofs TMA et al.: Adrenocortical carcinoma: A population-based study on incidence and survival in the Netherlands since 1993. Eur J Cancer 2013; 49(11): 2579-86 15 Fassnacht M et al.: Adrenocortical carcinoma: A clinician’s update. Nat Rev Endocrinol 2011; 7(6): 323-35 16 European Medicines Agency: EU/3/02/102: Orphan designation for the treatment of adrenal cortical carcinoma. https://www.ema.europa.eu/en/medicines/human/orphandesignations/eu302102 ; zuletzt aufgerufen am 11.05.2022 17 Terzolo M et al.: Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007; 356(23): 2372-80 18 Bedrose S et al.: Adjuvant therapy in adrenocortical carcinoma: Reflections and future directions. Cancers 2020; 12: 508
Das könnte Sie auch interessieren:

Prof. Mani Menon – Roboterchirurgie: vom Zweifel zum Durchbruch
Prof. Mani Menon spricht im ÖGU-Podcast mit Univ.-Prof. Shahrokh F. Shariat über die Anfänge der robotischen Chirurgie, prägende Rückschläge, Mentoren, Mut und Menschlichkeit in der Medizin.

„Buried penis“: Herausforderungen in der rekonstruktiven Urologie
Die rekonstruktive Urologie erfordert zunehmend individualisierte operative Konzepte bei komplexen anatomischen und funktionellen Ausgangssituationen. Der „buried penis“ stellt dabei ...

PCNL-Indikationen in Zeiten von flexibler Ureterorenoskopie mit Absaugung?
In der Behandlung von Nierensteinen, die größer als 2cm sind, spielt die perkutane Nephrolithotomie (PCNL) weiterhin eine wichtige Rolle. Die flexible Ureterorenoskopie (URS) mit ...