
Amyloidose und ihre orthopädischen Manifestationen
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Die Amyloidose ist eine Störung, bei der es zur Ablagerung und Anhäufung fehlgefalteter Proteine (Amyloid) im Gewebe kommt, wasein oder mehrere Organsysteme beeinträchtigen und zu schwerwiegenden kardialen und neurologischen Erkrankungen führen kann. Aufgrund häufig auftretender muskuloskelettaler Manifestationen1 können Orthopäd:innen eine aktive Rolle in der Früherkennung und einem baldigen Behandlungsbeginn einnehmen, um den Krankheitsverlauf zu verlangsamen.
Keypoints
-
Die Amyloidose ist eine verheerende Multisystemerkrankung, bei der es zu Ablagerungen und Anhäufungen fehlgefalteter Proteine kommt.
-
Mit der Entwicklung neuartiger Therapeutika, die sich als wirksam in der Verlangsamung des Krankheitsverlaufs erwiesen haben, hat das Interesse an der frühzeitigen Erkennung und Überweisung zur Einleitung der Therapie zugenommen.
-
Orthopädische Manifestationen (Karpaltunnelsyndrom, schnellender Finger, spontane Ruptur der distalen Bizepssehne, Erkrankungen der Rotatorenmanschette, lumbale Spinalkanalstenose) sind häufig und treten in der Regel viele Jahre vor kardialen Symptomen und vor der formellen Diagnose der Amyloidose auf.
-
Orthopäd:innen befinden sich daher in der einzigartigen Position, ihren Patienten zu helfen, indem sie Red Flags erkennen, sofern erforderlich Biopsien durchführen und bei nachgewiesener Amyloidablagerung an Spezialist:innen überweisen.
Die beiden häufigsten Formen von Amyloidose sind die Transthyretin-Amyloidose (ATTR), bei der die Ablagerung von Wildtyp(-ATTRwt) oder mutiertem (ATTRm) Transthyretin erfolgt, und die Leichtketten-Amyloidose (AL), bei der fehlgefaltete monoklonale Immunglobulin-Kappa- oder -Lambda-Leichtketten abgelagert werden.2,3 Die ATTRm ist durch heterozygote Mutationen im TTR-Gen bedingt und tritt daher familiär gehäuft auf. Sie kann durch einen einfachen Gentest nachgewiesen werden.
Je nach Art der Amyloidose können verschiedene Organsysteme betroffen sein und die klinische Präsentation kann kardiale, neurologische, renale, pulmonale, hepatische, hämatologische und muskuloskelettale Manifestationen umfassen.2–4
Kardiale Symptome sind ein häufiges Endstadium der systemischen Amyloidose. Die Ablagerung von Amyloid im Herzmuskelgewebe führt zu einer infiltrativen restriktiven Kardiomyopathie, die in einer Herzinsuffizienz mit typischerweise erhaltener Ejektionsfraktion resultiert.2,3 Die Prognose von Patienten mit kardialer Amyloidbeteiligung ist besonders schlecht; die mittlere Überlebensrate beträgt 6 Monate für AL-Amyloidose-Patienten mit Herzinsuffizienz und 4 Jahre für ATTRwt-Amyloidose-Patienten.3 Die Ablagerung von Amyloid im peripheren und autonomen Nervensystem führt zu peripherer Neuropathie, orthostatischer Hypotonie und gastrointestinaler Dysmotilität.3,4
Es sind zahlreiche orthopädische Manifestationen der Amyloidose bekannt. Die Ablagerung von Amyloid in den Weichteilen des muskuloskelettalen Systems kann zu einem Karpaltunnelsyndrom, einem schnellenden Finger, einer traumatischen distalen Bizepssehnenruptur, einer Läsion der Rotatorenmanschette und einer lumbalen Spinalkanalstenose führen.2–6 Diese Erscheinungsbilder der systemischen Amyloidose sind häufig und treten oft 5–10 Jahre vor kardialen Symptomen auf.2 Daher kann das Erkennen von Red Flags eine frühzeitige Diagnose ermöglichen.
Neuartige Therapien zur Behandlung von Amyloidose
Es konnten neuartige therapeutische Wirkstoffe für die Behandlung der hereditären ATTRm-Amyloidose entwickelt werden. Die Transthyretinsynthese findet hauptsächlich in der Leber statt. Die Medikamente Patisiran, Vutrisiran und Inotersen sollen diesen Vorgang hemmen, was zu einer verringerten Produktion und Anhäufung fehlgefalteter Amyloidproteine im Gewebe führt. Unter Therapie konnte eine Verbesserung des neurologischen Verlaufs und der Lebensqualität bei Patienten mit ATTRm festgestellt werden.7,8 Die Wirkstoffe Diflunisal und Tafamidis wirken durch Silisierung der tetrameren Form von Transthyretin, indem sie dessen Dissoziation in die monomere Form hemmen, welche die fehlgefalteten Amyloidproteine ausfällt. Diflunisal konnte neurologische Beeinträchtigungen reduzieren, während Tafamidis (das nun auch bei der ATTRwt mit kardialer Beteiligung zugelassen ist) bei Patienten mit kardiomyopathischer ATTR-Amyloidose die Mortalität und kardiovaskuläre Hospitalisierungen verringerte. Unter beiden Arzneimitteln kam es zu einer Verbesserung der Lebensqualität.9,10
Patienten mit AL-Amyloidose, die Symptome einer Herzinsuffizienz aufweisen, werden mit einer Dreifach-Medikamententherapie bestehend aus Bortezomib, Dexamethason und Cyclophosphamid behandelt. Die mittlere Überlebenszeit kann somit auf über 2 Jahre verlängert werden, während sie unbehandelt bei weniger als 6 Monaten liegt.11 Bei Patienten mit AL-Amyloidose ohne schwerwiegende kardiale Beteiligung kann eine Knochenmarktransplantation in Betracht gezogen werden.3
Die jüngste Entwicklung neuartiger Therapeutika, insbesondere für die ATTRm, ist sehr ermutigend. Sie können den natürlichen Verlauf dieser verheerenden systemischen Erkrankung positiv beeinflussen. Allerdings konzentrieren sich die Wirkmechanismen in erster Linie darauf, die Progression der Ablagerungen zu verlangsamen. Die Prognose fortgeschrittener systemischer Amyloidose bleibt schlecht. Daher sind eine frühzeitige Diagnose und Einleitung einer Therapie sehr wichtig; sie können potenziell den klinischen Verlauf von Patienten mit Amyloidose beeinflussen.
Karpaltunnelsyndrom (CTS)
Das CTS ist die früheste und häufigste nichtkardiale Manifestation der systemischen Amyloidose.5 Ursächlich soll eine Verdickung und Verhärtung der Beugesehnenscheiden sein, wodurch der Nervus medianus gegen den querverlaufenden Karpaltunnel gedrückt wird.12 Ob das mit Amyloidose assoziierte CTS aufgrund von Amyloidablagerungen in den Beugesehnenscheiden und dem darüber liegenden querverlaufenden Handgelenksband entsteht, durch Ablagerungen im Nerv selbst oder durch beides, ist noch ungeklärt.3
Die Prävalenz des symptomatischen CTS bei Patienten mit ATTR-Amyloidose liegt bei 68%. Es ist der häufigste erste Befund bei 55% der Patienten mit ATTR-Amyloidose, gefolgt von Herzinsuffizienz bei 45%.3,13 Beschwerden treten in der Regel vor kardialen Symptomen auf und die eigentliche Diagnose der Amyloidose findet bei diesen Patienten im Durchschnitt 4–7 Jahre früher statt.3,13,14
Im Rahmen der chirurgischen Behandlung des CTS, der Karpaltunnelspaltung, kann intraoperativ eine Biopsie der benachbarten Beugesehnenscheide entnommen werden, um die Diagnose der Amyloidose zu bestätigen. Da das CTS die häufigste Kompressionsneuropathie der oberen Extremität ist und oft von orthopädischen Chirurg:innen behandelt wird, besteht so die Möglichkeit der frühzeitigen Erkennung der Amyloidose und Einleitung einer Behandlung.
Eine routinemäßige nichtselektive Biopsie bei Patienten mit idiopathischem CTS weist relativ wenige positive Ergebnisse für Amyloidose auf.15 Beschriebene Risikofaktoren für den Nachweis von Amyloid im Rahmen einer Karpaltunnelspaltung sind männliches Geschlecht und höheres Alter.16, 17 Weitere mit einer positiven Biopsie assoziierte Faktoren umfassen das CTS-Rezidiv,18 bilaterales CTS und Red-Flag-Komorbiditäten wie Polyneuropathie, lumbale Spinalkanalstenose, spontane distale Bizepssehnenruptur, Herzinsuffizienz und Herzrhythmusstörungen.3
Schnellender Finger
Der schnellende Finger entsteht durch eine mechanische Diskrepanz zwischen der Größe der Beugesehnen und des A1-Ringbands, durch das sie verlaufen. Er kann jeden Finger betreffen und manifestiert sich klinisch als Schmerz in der Handfläche im Bereich des A1-Ringbands. In fortgeschrittenen Stadien kommt es zu mechanischen Blockaden sowie zu einem Schnellen des Fingers bei Beugung und Streckung.19 Es wird angenommen, dass sich Amyloid in den Beugesehnenscheiden ablagert. Die chirurgische Behandlung besteht in der A1-Ringbandspaltung und in der Tendosynovektomie im Sinne einer Abtragung des grün-bräunlichen Materials auf der Sehnenscheide als Zeichen des infiltrativen Krankheitsprozesses. Im Rahmen einer Untersuchung von Patienten mit idiopathischen Sehnenscheidenentzündungen im Bereich der Finger konnten bei 65% Amyloidablagerungen festgestellt werden. Diese Patienten wiesen im Durchschnitt eine höhere Anzahl betroffener Finger im Vergleich zu Patienten ohne Amyloidablagerungen auf.20
Orthopäd:innen sollten besonders auf die mögliche Diagnose der Amyloidose bei älteren Patienten mit schnellenden Fingern, die beidseitig auftreten oder mehrere Finger betreffen, sowie bei Patienten mit vorheriger oder gleichzeitiger Behandlung eines CTS achten.19
Distale Bizepssehnenruptur
Spontane Rupturen der distalen Bizepssehne treten im Rahmen eines minimalen oder belanglosen Traumas auf. Bei älteren Menschen können sie mit einer Ablagerung von Amyloid in der distalen Bizepssehne in Verbindung stehen, was zu einer Schwächung des Gewebes führt.21 In einem retrospektiven Vergleich der Häufigkeit von distalen Bizepssehnenrupturen unter Patienten mit Herzinsuffizienz aufgrund einer ATTRwt-Kardiomyopathie mit der von Patienten mit Herzinsuffizienz anderer Ursachen konnte eine deutlich höhere Inzidenz bei Patienten mit ATTRwT-Kardiomyopathie nachgewiesen werden (33,3% gegenüber 2,5%). In der ATTRwt-Gruppe trat die distale Bizepssehnenruptur im Schnitt 5 Jahre vor der Diagnose der Herzinsuffizienz auf.22 Der Befund einer spontanen Ruptur der distalen Bizepssehne sollte den Verdacht auf eine mögliche zugrunde liegende Amyloidose wecken, insbesondere bei älteren männlichen Patienten.
Läsionen der Rotatorenmanschette
Das Schultergelenk ist häufig von Amyloidose betroffen. Bei einem Drittel bis zur Hälfte der Patienten, die langfristig eine Hämodialyse erhalten, kommt es zu Schmerzen und Steifheit der Schulter. Die Beschwerden sind auf die progressive Ablagerung von Amyloid im Rahmen einer Amyloidose zurückzuführen.5,23 Bei fortgeschrittener Erkrankung kommt es zu Ablagerungen in der Rotatorenmanschette und im periartikulären Bindegewebe. Dies kann einerseits zu atraumatischen oder minimal traumatischen Rupturen, andererseits zu einer Weichteilschwellung im anterioren Schulterbereich, die als „Shoulder-Pad-Sign“ bezeichnet wird, führen.23
Sonografische Befunde einer Amyloidose im Bereich des Schultergelenks umfassen eine Verdickung der Rotatorenmanschette, der Synovialscheide um die lange Bizepssehne und der Bursa subdeltoidea. Eine Verdickung der Sehne des Musculus supraspinatus von mehr als 7mm im Ultraschall gilt als diagnostisch für eine Schulteramyloidose.24,25 In der Magnetresonanztomografie (MRT) zeigt sich die Amyloidose der Schulter frühzeitig durch eine Verdeckung des Rotatorenintervalls und des subkorakoidalen Raums durch Fasergewebe sowie durch eine relative Verdickung der Rotatorenmanschette. In fortgeschrittenen Stadien können massive Rupturen der Rotatorenmanschette und knotige Bindegewebsbildungen subdeltoidal und im Recessus axillaris sichtbar werden. Ein MRT-basiertes Klassifikationssystem für die Hämodialyse-assoziierte Amyloidose der Schulter wurde entwickelt (Tab. 1).26

Tab. 1: MRT-Klassifikation der Amyloidose der Schulter (nach Ando et al. 2017)26
Eine chirurgische Behandlung der Manifestation einer Amyloidose in der Schulter ist erst nach dem Versagen konservativer Maßnahmen (NSAR, Physiotherapie, Cortison-Infiltrationen) angezeigt. Sie besteht aus arthroskopischer Synovektomie und Débridement des verdickten Synovialgewebes sowie bei Bedarf einer Rotatorenmanschettenrekonstruktion. Im fortgeschrittenen Stadium können eine offene Synovektomie mit Débridement oder die Implantation einer inversen totalen Schulterendoprothese in Betracht gezogen werden.26 Die für die Amyloidose typische synoviale Proliferation kann die arthroskopische Visualisierung beeinträchtigen und das Verfahren technisch anspruchsvoller machen.
Lumbale Spinalkanalstenose
Die lumbale Spinalkanalstenose resultiert aus der Kompression neuraler Elemente im Wirbelkanal. Einengungen der knöchernen und diskoligamentären Strukturen treten mit dem Alter und auch in Verbindung mit Instabilität oder Skoliose auf. Bei symptomatischer Ausprägung kann die lumbale Spinalkanalstenose radikuläre Beschwerden, neurogene Claudicatio oder ein Cauda-equina-Syndrom verursachen.
Über die Beziehung von Amyloidose zur lumbalen Spinalkanalstenose ist wenig bekannt und Biopsien werden nicht routinemäßig durchgeführt. Es existiert eine allgemeine Annahme, dass Amyloid im Ligamentum flavum abgelagert wird und so zu dessen Hypertrophie führt.27–31 Dies ist eine häufig unerkannte und unterdiagnostizierte zugrunde liegende Pathologie, die bei 13%–44% aller Patienten mit Spinalkanalstenose beschrieben wird. Eine histologisch und biochemisch bestätigte Amyloidablagerung kann zur frühzeitigen Diagnose und Behandlung beitragen.31
Die klinische Bedeutung mikroskopisch identifizierbarer Amyloid-Infiltrationen im Ligamentum flavum ist unklar, aber größere Transthyretin-positive Ablagerungen könnten pathogenetisch sein. Möglicherweise besteht eine Korrelation zwischen der Menge des Amyloids und der Dicke des Ligamentum flavum. Die Nützlichkeit von Biopsien des Ligamentum flavum für die frühzeitige Erkennung von Amyloidose ist bis dato nicht geklärt. Eine Biopsie sollte beispielsweise bei abnormen Nervenleitungsmessungen in Betracht gezogen werden oder bei sich verschlechternden Symptomen trotz chirurgischer Behandlung.32
Zukünftige Studien könnten darauf abzielen, neuartige Methoden zur nichtinvasiven Identifizierung von Amyloid zu entwickeln. Die 99mTc-PYP-Szintigrafie könnte hierfür eine Option darstellen. Obwohl diese Art der nuklearen Szintigrafie traditionell für die Diagnose von ATTRwt-Kardiomyopathie verwendet wurde, deuten einige Fallberichte darauf hin, dass PYP-Aufnahme auch in anderen Geweben jenseits des Myokards stattfindet. Zukünftige Studien sind erforderlich, um zu zeigen, ob dieser Vorgang auch im Bereich der Wirbelsäule möglich ist und ob dies mit ATTRwt-Ablagerungen im Gewebe korreliert.31
Die Rolle der Orthopäd:innen: Möglichkeiten der Früherkennung
Das gesteigerte Interesse an der frühzeitigen Erkennung und Überweisung zur Einleitung der Therapie bei Amyloidose resultiert aus der Einführung neuartiger Medikamente, die ihre Wirksamkeit insbesondere bei der Verlangsamung des Krankheitsverlaufs bewiesen haben. Orthopädische Manifestationen treten häufig viele Jahre vor kardialen Symptomen und der formellen Diagnosestellung der Amyloidose auf. Daher sind Orthopäd:innen in einer einzigartigen Position, ihren Patienten zu helfen. Dies kann durch das Erkennen von Red Flags, das Durchführen von Biopsien bei Bedarf und die Überweisung im Falle nachgewiesener Amyloidablagerungen an Spezialist:innen erfolgen (Abb. 1).
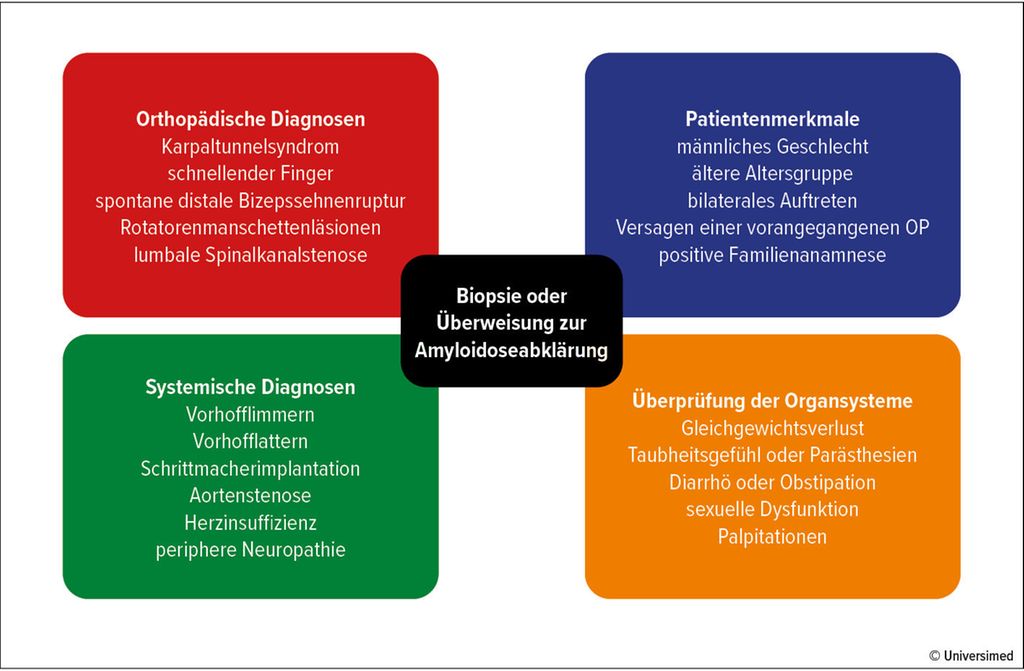
Abb. 1: Red Flags, Merkmale und Diagnosen, die Orthopäd:innen dazu veranlassen können, eine Biopsie durchzuführen oder eine Überweisung wegen Amyloidose vorzunehmen
Besonders bei älteren und männlichen Patienten sollten Behandelnde wachsam hinsichtlich der möglichen Diagnose einer Amyloidose sein. Wichtige Aspekte der Krankengeschichte können bilaterale Symptome, das Versagen vorheriger Operationen und eine positive Familienanamnese umfassen. Eine Biopsie während routinemäßiger orthopädischer Verfahren sollte für Patienten mit Red Flags für systemische Amyloidose in Betracht gezogen werden.3,32,33
Aktuelle Wissenslücken umfassen unabhängige Faktoren, die mit einer positiven Biopsie für die Ablagerung von Amyloid in jedem orthopädischen Verfahren außer der Karpaltunnelspaltung assoziiert sind. Zukünftige qualitativ hochwertige Studien sind erforderlich, um diese Faktoren zu identifizieren. Es gilt, Risikoscores für positive Biopsien zu entwickeln, um die Risikoeinstufung steuern zu können.
Literatur:
1 Zhang D et al.: J Am Acad Orthop Surg 2021; 29(10): 488-96 2 Cuddy SAM, Falk RH: Can J Cardiol 2020; 36: 396-407 3 Donnelly JP et al.: J Hand Surg Am 2019; 44: 868-76 4 Siddiqi OK, Ruberg FL: Trends Cardiovasc Med 2018; 28: 10-21 5 Kurer MH et al.: J Bone Joint Surg Br 1991; 73: 271-6 6 Sekijima Y et al.: Amyloid 2018; 25: 8-10 7 Adams D et al.: NEngl J Med 2018; 379: 11-21 8 Benson MD et al.: N Engl J Med 2018; 379: 22-31 9 Berk JL et al.: JAMA 2013; 310: 2658-67 10 Maurer MS et al: N Engl J Med 2018; 379: 1007-16 11 Sperry BW et al: J Am Coll Cardiol 2016; 67: 2941-8 12 Grokoest AW, Demartini FE: JAMA 1954; 155: 635-7 13 Nakagawa M et al.: Amyloid 2016; 23: 58-63 14 Papoutsidakis N et al.: J Card Fail 2018; 24: 131-3 15 Bastian FO: Am J Clin Pathol 1974; 61: 711-7 16 Sekijima Y et al.: Hum Pathol 2011; 42: 1785-91 17 Sperry BW et al.: JAm Coll Cardiol 2018; 72: 2040-50 18 Scott KL et al.: Plast Reconstr Surg 2019; 143: 169-75 19 Ryzewicz M, Wolf JM: J Hand Surg Am 2006; 31: 135-46 20 Hara Y et al.: JHand Surg Asian Pac Vol 2020; 25: 340-4 21 Kocabas U, Önsel Türk U: Balkan Med J 2020; 37: 353-4 22 Geller HI et al.: JAMA 2017; 318: 962-3 23 Katz GA et al.: N Engl J Med 1973; 288: 354-5 24 Cardinal E et al.: Am J Roentgenol 1996; 166: 153-6 25 Sommer R et al.: J Ultrasound Med 2000; 19: 765-70 26 Ando A et al.: Knee Surg Sports Traumatol Arthrosc 2017; 25: 2217-24 27 Dowd RS et al.: World Neurosurg 2019; 131: 104-7 28 Aus dem Siepen F et al.: Clin Res Cardiol 2019; 108: 1324-30 29 Westermark P et al.: Ups J Med Sci 2014; 119: 223-8 30 Yanagisawa A et al.: Mod Pathol 2015; 28: 201-7 31 Wang AY et al.: World Neurosurg 2023; 177: 88-97 32 Adams D et al.: J Neurol 2021; 268: 2109-22 33 Ruberg F et al.: J Am Coll Cardiol 2019; 73: 2872-91