
Hypersensitivitätspneumonitis
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Eine neue Terminologie, ein spezifischer Fragebogen zur Abklärung von Verdachtsfällen in der Schweiz und begrenzte pharmakologische Behandlungsoptionen, das waren die Neuigkeiten zur Hypersensitivitätspneumonitis am diesjährigen SGP-Kongress in Luzern.
Die Hypersensitivitätspneumonitis (HP) macht etwa 10% aller interstitiellen Lungenerkrankungen (ILD) aus. In der Schweiz sind ca. 200 Personen von einer chronisch fibrosierenden oder einer nichtfibrosierenden HP betroffen. Deutlich höher ist die Zahl der Personen mit einer akuten HP. Während die akute HP bei vorgängig sensibilisierten Personen durch eine (in der Regel) mehrstündige Exposition gegenüber moderaten bis hohen Allergenmengen ausgelöst wird, entwickelt sich die chronische HP als Folge einer über Wochen bis Jahre andauernden niedrigen Allergenexposition.1 Ältere Studienergebnisse deuteten auf eine vorteilhaftere Prognose der chronischen HP im Vergleich zu anderen ILD hin.2 Neuere Daten zeigen nun, dass die Lebenserwartung vor allem vom Vorliegen einer parenchymalen Fibrose abhängig ist. So betrug die mediane Überlebensdauer bei Patienten mit nichtfibrosierender HP rund 15 Jahre, während sie bei Patienten mit fibrosierender HP bei 8 Jahren lag. Bei einer fibrosierenden HP mit «honeycombing» beträgt die mediane Überlebensdauer circa 2 bis 3 Jahre und ist damit mit derjenigen bei einer idiopathischen Lungenfibrose (IPF) vergleichbar.3 Die neuen Studiendaten haben die Terminologie in den aktuellen HP-Guidelines beeinflusst: Anstelle der bisherigen Stratifikation in akute, subakute und chronische HP wird die HP neu in fibrosierende und nichtfibrosierende HP unterteilt.
Multidisziplinäre Diagnosestellung von Vorteil
Die Diagnose, insbesondere der fibrosierenden HP, gestaltet sich aufgrund der Symptomvielfalt und der zahlreichen Überlappungen mit anderen ILD als schwierig. Wichtig ist die Erfassung von HP-relevanten Triggern mittels Anamnese und strukturierter Patientenbefragungen. Eine Delphi-Umfrage bei Experten hat eine Liste mit den wichtigen Auslösern generiert (Abb. 1).4 Zur Abklärung von Verdachtsfällen in der Schweiz hat die «Special Interest Group Interstital And Rare Lung Diseases» der Schweizerischen Gesellschaft für Pneumologie einen Fragebogen mit relevanten Triggern entworfen, der in mehreren Sprachen zur Verfügung steht. Darüber hinaus liefern die HRCT («high-resolution computed tomography») und die Bronchoskopie, inkl. Lungenbiopsie und bronchoalveolärer Lavage (BAL), wichtige Hinweise. Häufig wird auch die Bestimmung spezifischer IgG-Antikörper (Präzipitine) diskutiert. Die Guidelines empfehlen ein solches Vorgehen wegen der geringen Spezifität nicht. «Der Nachweis von Präzipitinen kann die Diagnose unterstützen, ist aber alleine nicht ausreichend für die Diagnose einer HP», sagte PD Dr. med. Sabina Guler vom Universitätsspital, Inselspital in Bern. Die Durchführung eines bronchialen Provokationstests im Rahmen der Routinediagnostik wird ebenfalls nicht empfohlen.
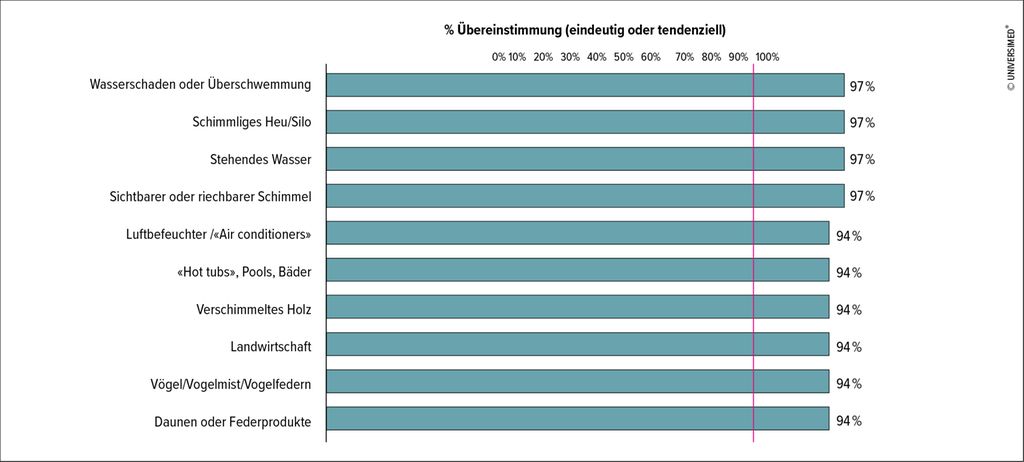
Abb. 1: Welche Expositionen sind relevant? (Adaptiert nach Barnes et al.)4
«Die Diagnose wird idealerweise im Rahmen einer multidisziplinären Diskussion unter Einbezug der verschiedenen Puzzleteile gestellt», sagte die Spezialistin. Steht kein multidisziplinäres Team zur Verfügung, kann ein Algorithmus mit diagnostischen Kriterien bei der Diagnose hilfreich sein.5 Der Algorithmus zeigt bei einer Diagnosesicherheit von ≥70% eine gute Performance (Sensitivität: 0,74; Spezifität: 0,9). Im Vergleich zum multidisziplinären Team war der Algorithmus hinsichtlich der Diagnosesicherheit aber unterlegen, wie eine Studie der Referentin zeigte.6 «Die multidisziplinäre Diagnose ist im Moment das Beste, was wir haben», so Guler.
Fehlende Therapieguidelines
Bislang gibt es keine offiziellen Behandlungsempfehlungen für die HP. Zudem existieren nur wenige klinische Studien, die eine pharmakologische Behandlung bei HP untersuchen. «Eine wichtige Massnahme ist die Entfernung des Triggers», sagte PD Dr. med. Christian Clarenbach vom Universitätsspital Zürich. Diese führe bei Patienten mit aktiver HP zu einer sichtbaren Erholung des Lungengewebes sowie zu einer verbesserten respiratorischen Funktion und unterstütze die Diagnose. Einen deutlich geringeren bis gar keinen Effekt haben die Karenzmassnahmen bei Patienten mit chronisch fibrosierender HP.
Immunsuppressive Therapien
Die Untersuchungen mit Immunsuppressiva zeigen kontroverse Ergebnisse. Zur Behandlung mit Kortikosteroiden fand sich lediglich eine kleine randomisierte Studie aus dem Jahr 1992 mit 36 Patienten mit einer aktiven HP.7 Diese waren für die Dauer von 8 Wochen entweder mit Prednisolon 5mg täglich oder Placebo behandelt worden. Wie die Ergebnisse einen Monat nach dem Behandlungsbeginn zeigten, erholte sich die Lungenfunktion unter der Kortikosteroidtherapie schneller als unter Placebo.
Zwei retrospektive Untersuchungen evaluierten den Effekt einer immunsuppressiven Therapie mit Mycophenolat mofetil (MMF) oder Azathioprin (AZA). In die erste Studie waren Patienten mit chronischer HP und einer Basistherapie mit Prednison (12mg täglich) eingeschlossen. Nach einer medianen Behandlungsdauer von einem Jahr zeigte sich keine eindeutige Zunahme der Vitalkapazität (FVC) durch die zusätzliche Behandlung mit MMF oder AZA. Die Diffusionskapazität (DLCO) hatte sich dagegen unter der Behandlung signifikant um 4,2% verbessert p<0,001).8 Bei der zweiten Untersuchung handelte es sich mehrheitlich um Patienten mit chronischer HP, die entweder mit Prednison, MFF oder AZA behandelt wurden. Dabei zeigte sich hinsichtlich der FVC kein Unterschied zwischen den verglichenen Behandlungen.9Aufhorchen lässt, dass die Patienten in dieser Studie, die nicht mit Immunsuppressiva behandelt worden waren, länger lebten als solche mit einer immunsuppressiven Therapie.
Eine Behandlungsoption könnte auch der Anti-CD20-Antikörper Rituximab sein, der mit Erfolg zur Behandlung von Kollagenose-assoziierten ILD eingesetzt wird. Die bislang verfügbaren zwei Studien mit Rituximab bei HP sind aufgrund der niedrigen Patientenzahlen (n<10) allerdings zu wenig aussagekräftig, um daraus eine Therapieempfehlung ableiten zu können.
Antifibrotische Therapie
In den letzten Jahren sind verschiedene Studien erschienen, die den Einfluss einer antifibrotischen Therapie mit Nintedanib bei ILD-Patienten untersucht haben. Nachdem die Studien INPULSIS-1 und -2 bei Patienten mit IPF etwa eine Halbierung der FVC-Abnahme im Vergleich zu Placebo gezeigt hatten, konnte ein vergleichbarer Effekt auch in den Studien INBUILD-1 und -2 bei Patienten mit fibrosierender ILD und in der SENSCIS-Studie bei Sklerodermie-assoziierter ILD gezeigt werden.11 Eine Subgruppenanalyse der INBUILD-Studien zeigte, dass die Behandlung mit Nintedanib auch bei Patienten mit chronisch fibrosierender HP zu einer verzögerten Abnahme der FVC führt.12
Lungentransplantation
«Für jüngere HP-Patienten ohne relevante Komorbiditäten, die nicht auf die pharmakologische Therapie ansprechen und bei denen die Lungenfunktion kontinuierlich weiter abnimmt, könnte die Lungentransplantation eine Therapieoption sein», so Clarenbach. Die häufigsten Indikationen für eine Lungentransplantation sind die IPF mit 30%, gefolgt von der COPD mit 26% und der zystischen Fibrose mit 13%. Wichtig sei eine frühzeitige Zuweisung von Patienten mit ILD.
Quelle:
Jahreskongress der Schweizerischen Gesellschaft für Pneumologie, 30. März bis 1. April 2022, Luzern
Literatur:
1 Costabel U et al.: Hypersensitivity pneumonitis. Nat Rev Dis Primers 2020; 6: 65 2 Ryerson CJ et al.: Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014; 145: 723-8 3 Salisbury ML et al.: Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest 2019; 155: 699-711 4 Barnes H et al.: A systematically derived exposure assessment instrument for chronic hypersensitivity pneumonitis. Chest 2020; 157: 1506-12 5 Morisset J et al.: Identification of diagnostic criteria for chronic hypersensitivity pneumonitis: an international modified delphi survey. Am J Resp Crit Care Med 2018; 197: 1036-44 6 Guler SA et al.: Performance of a diagnostic algorithm for fibrotic hypersensitivity pneumonitis. A case-control study. Respir Res 2021; 22: 120 7 Kokkarinen JI et al.: Effect of corticosteroid treatment on the recovery of pulmonary function in farmer‘s lung. Am Rev Respir Dis 1992; 145: 3-5 8 Morisset J et al.: Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest 2017; 151: 619-25 9 Adegunsoye A et al.: Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res 2017; 3: 00016-2017 10 Morell F et al.: Addition of rituximab to oral corticosteroids in the treatment of chronic hypersensitivity pneumonitis. Arch Bronconeumol 2020; 56: 255-6 11 Bonella F et al.: Meta-Analysis of effect of nintedanib on reducing FVC decline across interstitial lung diseases. Adv Ther 2022; 39: 3392-402 12 Wells AU et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 2020; 8: 453-60
Das könnte Sie auch interessieren:

Asthma und der zirkadiane Rhythmus
Der zirkadiane Rhythmus spielt nicht nur beim Schlafverhalten eine bedeutende Rolle, sondern hat auch einen erheblichen Einfluss auf Asthmaanfälle und die Lungenfunktion. Die gezielte ...


Hypersensitivitätspneumonitis – wie oft denken wir Pathologen nicht daran?
Die Hypersensitivitätspneumonitis (HP) ist eine immunvermittelte interstitielle Lungenerkrankung, die durch Immunreaktionen auf inhalierte Antigene verursacht wird. Die Diagnose stützt ...